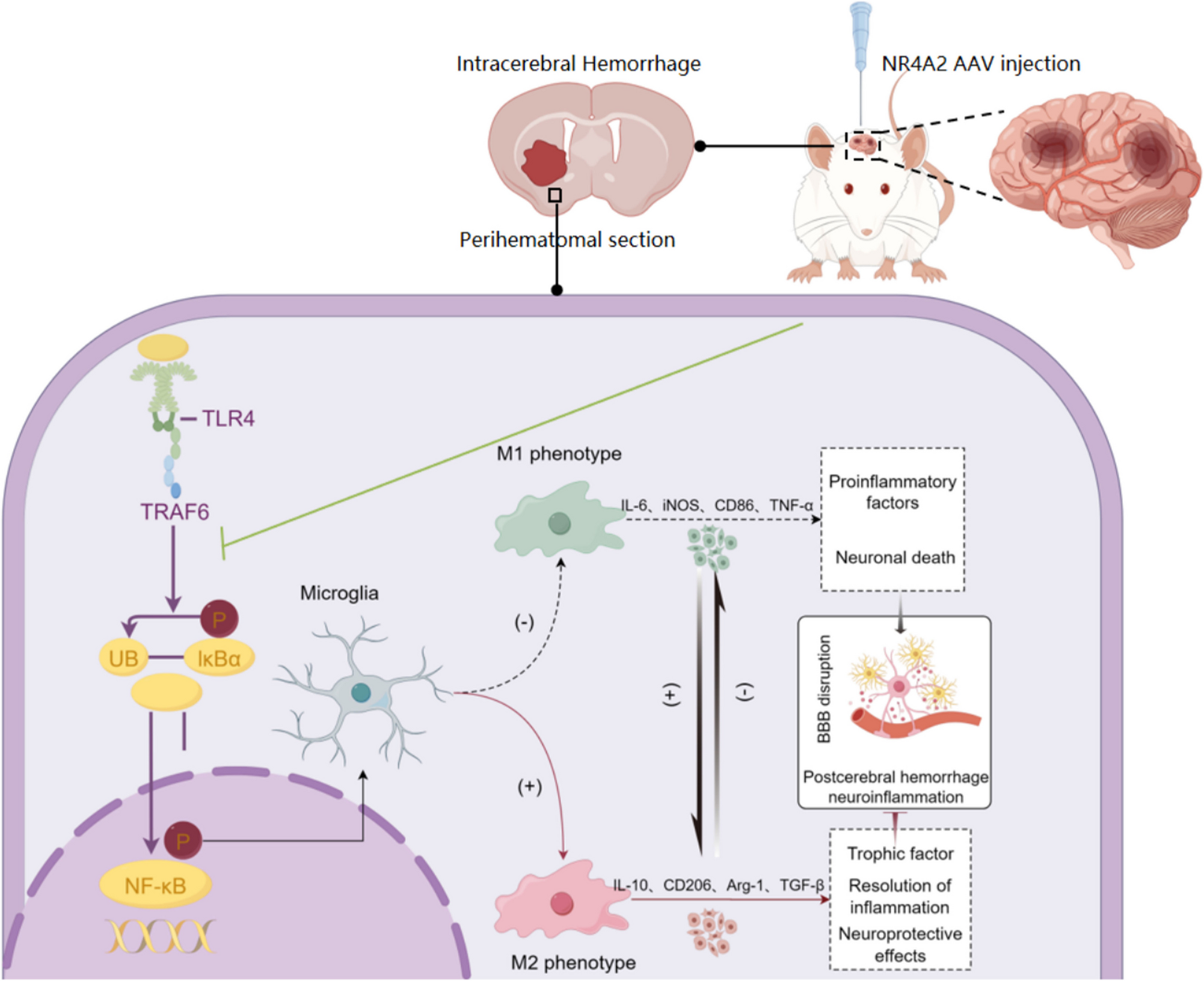
Focal ICH model and drug treatment
Sprague–Dawley (SD) rats (220–250 g) were obtained from Guangzhou Bestong Biotechnology Co., Ltd., China, and the experimental scheme for the rats was reviewed and approved by the Animal Protection and Utilization Committee of the First Affiliated Hospital of Guangzhou Medical University. The rats were acclimated to the laboratory environment for at least 1 week before the experiment, with a 12-h light/dark cycle and free access to food and water. The collagenase-induced ICH model in rats was established as previously described. Collagenase VII (Sigma-C0773, 2 μl sterile saline, 0.6 U) was prepared in advance and stored on ice during use. After anesthetizing the rats with isoflurane, the prepared collagenase VII was injected into the striatum at the following position relative to the striatum: anteroposterior, −3 mm; lateral, 1 mm; and ventral, 6 mm. After the injection, the syringe was kept in place for another 5–10 min to ensure even diffusion of the collagenase, after which the syringe was slowly withdrawn. After the operation, the rats were placed in a warm recovery box, and their vital signs were monitored until they fully recovered from anesthesia. The rats were provided free access to food and water after they woke. The rats were observed for any abnormal behaviors, such as limb paralysis, abnormal posture, or decreased activity level [27]. The TLR4 agonist CRX-527 (0.25 mg/kg; #tlrl-crx527, InvivoGen) was dissolved in DMSO and then intraperitoneally injected before ICH injury, as described in a previous study [28].
Cell culture and reagents
The brain microvascular endothelial cells (bEnd.3) and monocytic cell line (THP-1) utilized in this study were procured from Procell Life Science & Technology Co., Ltd. These cells were used primarily to construct an in vitro BBB model and to investigate endothelial inflammatory infiltration under hemorrhagic conditions. bEnd.3 cells were cultured in DMEM supplemented with fetal bovine serum (FBS, NEST Biotechnology) in culture flasks (SAINING, Biotechnology) at 37 °C in a 5% CO2 incubator. In contrast, THP-1 cells were cultured in RPMI-1640 medium supplemented with 10% FBS, and the remaining culture conditions were identical to those used for bEnd.3 cells. The microglial cell line BV2 used in this project was a generous gift from Dr. Cheng Huang and was primarily used to study the polarization of microglia under in vitro hemorrhagic conditions, with culture conditions identical to those of bEnd.3 cells.
Forelimb force test
Forelimb force is an important indicator for evaluating motor capacity, muscle function, and motor function recovery in some models of disease or injury [29]. First, a device specifically designed to immobilize experimental rats should be designed to steadily limit the animal's body movement while ensuring that the forelimb can move freely and make contact with the force sensor. The acclimated and trained animal is then carefully placed in the animal holding device. The stimulation and induction device is activated to provide the animal with a specific stimulus to induce forceful forelimb movement. After each stimulation, the maximum and average forelimb force and the curve of force change over time were recorded. Each measurement was repeated several times (e.g., 10–15 times), and an appropriate rest time (e.g., 30–60 s) was given between each measurement to avoid animal fatigue affecting the results of the experiment. Finally, the maximum, average and standard differences in forelimb force measured many times for each animal were calculated and analyzed.
Rotarod test
The rotarod test is used to evaluate motor coordination, balance, motor learning and memory in animals and can provide data support for the study of neurological diseases, drug efficacy, and the repair of sports injuries [29]. Before the experiment, the animals were maintained in a standard environment for at least 3–5 d to acclimate them to the new environment. On the day of the experiment, the animals were moved to the test environment 1–2 h in advance so that they could acclimate to the test site. The animal was placed gently on the swivel, the swivel was activated, and the timer was started. The time from the time the animal was placed on the rod to the time it was dropped, that is, the residence time, was recorded. Each animal was tested 3–5 times, each time with an interval of 10–15 min, and the average residence time was taken as the test result of the animal at this rotational speed. Data such as the residence time and drop speed obtained from each test are recorded in a spreadsheet, and abnormal data are checked and removed.
Nissl staining
The freshly obtained animal tissues were promptly immersed in a 4% paraformaldehyde fixation solution and rinsed three times with 0.1 M PBS, with each rinse lasting 15 min. Thereafter, the tissue was placed in a cryostat microtome and sectioned into slices with a thickness of 10–20 μm. The slide was then immersed in a 1% toluidine blue staining solution (purchased from Yesaen Biotechnology; product number 60531ES50) and incubated at 37 °C for 15–30 min to enable the Nissl bodies to be fully stained. After differentiation, the slices were cleared in xylene for 3–5 min. Subsequently, an appropriate amount of neutral balsam was dropped onto the slices, and a coverslip was carefully placed on top to avoid the formation of air bubbles. Once the mounting medium had completely dried, the samples were observed under an optical microscope.
HE staining
The paraffin sections were immersed in xylene I and xylene II successively, with each immersion lasting 10–15 min. After thorough dewaxing, the sections were carefully removed. The sections subsequently underwent a progressive hydration process in which various concentrations of alcohol were used. Thereafter, the hydrated sections were stained with hematoxylin dye solution (Beijing Solarbio) for 5–10 min. Following a gentle rinse with water, the samples were transferred to a 1% hydrochloric acid-alcohol solution (composed of 99 ml of 75% alcohol and 1 ml of concentrated hydrochloric acid) for differentiation. This step was carried out for several seconds to achieve a moderately stained nucleus. After the nuclei were properly stained and the sections were blue-toned, they were immersed in eosin dye solution for 3–5 min to stain the cytoplasm. Finally, by closely observing the alterations in the shape, color, and structure of the tissues and cells under a microscope, it became possible to determine whether any pathological changes were present accurately.
FITC-Dextran analysis
To analyze BBB permeability in ICH rats, FITC-dextran (10 kDa, 400 mg/kg) was injected into the basal ganglia of the rats via the tail vein. After 30 min, the blood vessels of the whole body were irrigated with 200 ml of normal saline to remove excess fluorescent substances in the blood vessels. The infiltration of FITC-dextran (10 kDa) into the brain section (transverse section) at the collagenase injection site was subsequently observed under a Carl Zeiss microscope.
Immunofluorescence staining and immunoblotting
Target genes, inflammatory factors, M1/M2 markers and tight junctions were detected via immunofluorescence (IF) staining. The whole protocol involved primary antibodies, including NR4A2 (Abcam; ab176184), CD86 (CST; #91882), iNOS (Abcam; ab283655), CD206 (CST; #24595), Arg-1 (CST: #93668), IBA-1 (Abcam; ab178847), GFAP (Abcam; ab7260), MPO (Abcam; ab208670), TLR4 (CST; #38519), ZO-1 (Abcam; ab307799), Claudin5 (Affinity; AF5216), TRAF6 (Abcam; ab40675), p-NF-κB p65 (CST; #3033), and NF-κB p65 (Signalway Antibody; 53227), and secondary antibodies. Images were captured via a Leica TCS SPII 5 confocal microscope. For immunoblotting, the denatured protein samples were separated from the upper and lower layers by dodecyl sulfate–polyacrylamide gel electrophoresis and gradually transferred to polyvinylidene fluoride membranes. The membrane was incubated in a dilute solution (Antibody diluent, Life-iLab, China) containing primary antibodies against NR4A2, CD86, iNOS, CD206, Arg-1, TLR4, TRAF6, and p-NF-κB p65, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Bioss USA). The immunoreactive bands were identified and captured with a gel imaging system.
Cell adhesion test
Once the growth density of bEnd.3 cells reached approximately 80%, an appropriate concentration of hemin was added, and a 24-h treatment was conducted. The cells were subsequently washed three times with PBS. The PKH26 reagent (BB-441125, Bestbio) was removed from the refrigerator in advance, after which it was prepared as a working solution. The THP-1 suspended cell precipitate was resuspended in 200 μl of the working solution, incubated in an incubator for 15 min, and then incubated in a 4 °C refrigerator for 30 min. The stained THP-1 cells were centrifuged, and the supernatant was discarded. The suspended cells were uniformly seeded onto the underlying bEnd.3 cells, incubated in an incubator for 3 h, and then, microscope images were taken.
Analysis of mRNA levels by real-time quantitative PCR
Total RNA was extracted via the TRIzol method. cDNA was synthesized via the SuperScript VILO cDNA Kit (Thermo Fisher Scientific, Inc.). The products were visualized with SYBR Green qPCR Master Mix (Applied Biosystems, USA). The primers used are listed in Table 1.
Table 1 The sequences of the primers in this studyRNA sequencing
Total RNA was isolated via TRIzol and enriched with oligo magnetic beads. The mRNA was randomly fragmented into small pieces by treatment with RNase III. Using the fragmented RNA as a template, reverse transcriptase and random primers are used to synthesize the first strand of cDNA, followed by the synthesis of the second strand. Construction of the cDNA library. Second-generation sequencing platforms are used to sequence the data. The sequencing data were used for gene expression quantification and alternative splicing analysis.
Adeno-associated virus (AAV) injection
The appropriate rats were selected and allowed to acclimate to the environment. sh-NR4A2 (NR4A2 knockdown) AAV, and NR4A2 (NR4A2 overexpression) AAV viruses with specific serotypes and titers were prepared. Anesthetize the rats. Then, the injection sites (anteroposterior, −1.5 mm; lateral, 3 mm; and ventral, 4.5 mm) were located, and the virus was slowly injected via a microsyringe. After the injection, the wound was sutured, and analgesics were administered. The rats were observed in their daily life. After two weeks, the expression of the target gene was detected at the molecular and cellular levels.
Chromatin immunoprecipitation (ChIP) assay
After digestion and extraction, the bEnd.3 cells were immersed in 1% formaldehyde and crosslinked on ice for 10 min. The crosslinking reaction was subsequently terminated by adding glycine to a final concentration of 125 mM and incubating the cells at room temperature for 5 min. Next, Bioruptor Plus was used to sonicate the chromatin, thereby generating DNA fragments. Then, the chromatin fragments were immunoprecipitated with antibodies against normal mouse IgG and NR4A2. After that, primers targeting the upstream and downstream regions of the target gene were designed, and the extracted DNA was used as a template. Finally, enrichment of the target gene region relative to the input control DNA was detected via quantitative PCR. The data were analyzed in detail by LC Biotech.
Pull-down assays
The bEnd.3 cells cultured in a 100-mm dish were harvested in 1 ml of cell lysis buffer (Yike Biotechnology Co., Ltd.) at 4 °C. The cell lysates were subsequently centrifuged at 12,000 × g for 20 min. The supernatant was preincubated with streptavidin-resin beads at 4 °C. Specifically, 10 μg of poly(dI-dC) was mixed with 1 ml of the precleared bEnd.3 cell lysates, which were diluted threefold, along with 0.5 μg of biotinylated double-stranded oligonucleotides. This mixture was incubated at 4 °C overnight. On the following d, an additional 25 μl of streptavidin beads was added, and the mixture was incubated for at least 1 h. The streptavidin beads were then pelleted via centrifugation at 5,000 × g for 10 min and washed with lysis buffer. Next, 30 μl of 2 × SDS buffer was added to elute the bound proteins. Finally, the presence of the NR4A2 protein was detected.
Dual-luciferase assays
The binding sites were predicted via the JASPAR database. KeyGen Biotech Co., Ltd. designed and synthesized the sequences of the wild-type (WT) and mutant (MUT) TLR4 mRNAs in the 3'-untranslated region (3'-UTR) and cloned them into a fluorescent reporter plasmid. Initially, bEnd.3 cells were transfected with plasmids. One plasmid encoded a firefly luciferase reporter gene regulated by the TLR4 promoter, whereas the other carried the Renilla luciferase gene, which served as a control for monitoring transfection efficiency. After a 6-h incubation, the growth medium was changed. The cells were subsequently treated with 80 μM hemin and incubated for another 24 h [30]. The luciferase and Renilla activities were then measured via the Promega Dual-Luciferase Reporter Assay System.
Evan blue permeability experiment
After the rats in each group were grouped and treated, 40 mg/kg Evans blue solution (Beyotime Biotechnology) was injected via the tail vein to ensure full administration. The animals were then placed in a warm, quiet environment for 24 h to allow solution circulation. The next day, brain tissues around the hematoma were extracted from each group. The collected tissues were ground, and an appropriate homogenate was taken and centrifuged at 5000 rpm for 10 min. DMSO was added to dissolve the Evans blue in the precipitate. The absorbance of the reconstituted samples was measured at 620 nm with a spectrophotometer, and the Evans blue content was calculated based on the standard curve.
Transmission electron microscopy
The tissues surrounding the rat brain hematoma were removed and immersed in a 3% glutaraldehyde fixing solution. After fixation, the tissue was rinsed with 0.1 mol/L sodium dimethylarsenate buffer, dehydrated under gradient conditions, permeated with acetone, and finally placed in an Eponate12 embedding agent at room temperature overnight. The embedding blocks were then placed on the microtome. Ultrathin slices with thicknesses of 40–60 nm were cut, transferred to copper grids, stored in a dry, light-protected environment and observed under an electron microscope.
In vivo coimmunoprecipitation assay
The surrounding hematoma tissue from each rat was obtained and cut into small pieces. RIPA buffer was added, the homogenate was centrifuged at 14,000 × g for 30 min at 4 °C, and the supernatant was collected. Specific antibodies were added to the supernatant. The mixture was incubated at 4 °C overnight. The protein A/G agarose beads (Biolinkedin, Shanghai, China) were washed three times and then added to the mixture above. The mixture was incubated at 4 °C for 2 h. The mixture was subsequently centrifuged, the supernatant was discarded, and the beads were washed 3 times. Loading buffer was added, and the protein was eluted by boiling in a water bath for 10 min. SDS-PAGE electrophoresis was performed, the proteins were transferred to a membrane, which was subsequently blocked. The samples were incubated with primary and secondary antibodies. The membrane was washed with TBST. The proteins were detected via a chemiluminescent substrate.
Statistical analysis
The results are presented as the means ± standard deviations. Each experiment was performed at least three times. Student's t-test was used for two groups, and ANOVA was used for three or more groups via GraphPad Prism 9. Statistical significance was determined as follows: *P < 0.05, ** P < 0.01, *** P < 0.001.



Comments (0)