Animal models
Gm5532 knockout (KO) mice (C57BL/6 J strain) were produced through CRISPR/Cas9-mediated excision of exons 1 and 2 (Shanghai Model Organisms, Shanghai, China). The mice were kept in a SPF animal facility (temperature, 23–25 °C; relative humidity, 40 ± 5%; light cycle: 12-h photoperiod), and received standard chow and distilled water, allowing ad libitum access. Each cage accommodated five mice.
The present study used 3 types of osteoporosis, namely, senile osteoporosis, postmenopausal osteoporosis and iron-overload induced osteoporosis.
Fifteen-month-old mice were used as aged mice. For postmenopausal osteoporosis, homozygous wild-type and Gm5532 KO female mice (5 weeks old) received bilateral ovariectomy (OVX) or sham procedure (sham). Samples of OVX and Sham mice were collected after 3 months. For iron-overload induced osteoporosis, homozygous wild-type and Gm5532 KO male mice (5 weeks old) were treated with intraperitoneal injections of 40 mg/kg ferric ammonium citrate (FAC) or equal volumes of physiological saline every three days for eight weeks. WT and KO mice were randomly grouped following simple randomization. No criteria were set to exclude animals during the experiments. At the indicated time points, cervical dislocation was performed to sacrifice the mice for collecting blood and femur samples. The left femur was harvested for micro-CT analysis. The right femur was harvested for histological analysis. Animal Ethics and Welfare Committee of Guizhou University of Traditional Chinese Medicine approved the procedures of animal experiments (20,220,169, 20,220,170, 20,220,170). All animal procedures were carried out in accordance with the ARRIVE guidelines.
Micro-computed tomography, histology, and ELISA assay
Femoral samples underwent fixation (10% formaldehyde) and subjected to micro-CT scanning (Bruker, Kontich, Belgium) at a resolution of 6.5 μm. For trabecular microarchitecture analysis, a 1-mm segment of distal trabecular bone situated 0.5 mm above the growth plate was designated as region of interest (ROI). After three-dimensional imaging, trabecular bone volume fraction, separation, number and thickness, and cortical bone volume and thickness were quantified.
For histological examination, femurs were processed by fixation in 10% formaldehyde, decalcification in 10% EDTA, and paraffin embedding. Paraffin sections of 2 μm thickness received TRAP staining to assess osteoclast numbers.
For ELISA analysis, blood samples were harvested at the indicated times and subjected to centrifuge (3000 rpm, 16 min, 4 °C). The concentrations of C-terminal telopeptide of type 1 collagen (CTX1) and tartrate-resistant acid phosphatase 5b (TRAP5b) were determined using the corresponding ELISA kits (Shanghai Meilian, Shanghai, China).
Cell culture and osteoclast differentiation
To obtain Bone marrow-derived macrophages (BMMs), femoral marrow cells were extracted from the male mice (5 weeks old) and cultured overnight (medium: α-MEM + 10% fetal bovine serum + 2 mM L-glutamine; conditions: 37 °C in 5% CO₂). Suspended cells were collected and were cultured with macrophage colony-stimulating factor (M-CSF; 25 ng/mL). For osteoclast differentiation, BMMs were plated at 10,000 cells per square centimeter in osteoclastogenic medium consisting of 25 ng/mL M-CSF plus 50 ng/mL receptor activator of NF-κB ligand (RANKL) for six days. Osteoclast identification was performed by TRAP staining and microscopic quantification of multinuclearity (threshold: 3 fused nuclei). Positive cells exhibited both TRAP + staining and characteristic multinuclear morphology. TRAP enzymatic activity was assessed using a pNPP-based kit (Beyotime, Shanghai, China). PeproTech (Rocky Hill, NJ, USA) provided M-CSF and RANKL.
RNA Sequencing (RNA-seq) and bioinformatics analysis
After treating cells for 3 days with the cytokine cocktail (RANKL + M-CSF), we performed TRIzol-based RNA isolation (Beyotime), and assessed RNA integrity (RIN > 8.0) via Bioanalyzer 2100. RNA samples were processed into sequencing libraries and run on the Illumina HiSeq system (Majorbio Biotech, Shanghai, China). Sequencing reads were quality-controlled and HISAT2-mapped to the Mus musculus reference genome. Transcript abundance was expressed as FPKM values, and differential gene expression was determined with DESeq2 (criteria: FDR < 0.05 and |log₂FC|≥ 1). Functional enrichment of differentially expressed genes was analyzed through GO and KEGG pathways using the clusterProfiler package.
Mitochondrial function analysis
After BMMs underwent 72-h differentiation, cells were stained with MitoTracker Red (Beyotime, 100 nM, 37 °C, 30 min) for cytometric analysis (BD, Franklin Lakes, NJ, USA). Relative ATP levels were quantified by monitoring NADH oxidation kinetics at 340 nm wavelength through UV spectrophotometry (Solarbio, Beijing, China). Mitochondria were isolated and used to measure the activities of complexes I-V with commercial kits (Solarbio). Using a Seahorse XFe24 Analyzer, sequential treatment with mitochondrial modulators (1.5 μM oligomycin → 2 μM FCCP → 0.5 μM rotenone/antimycin A) enabled OCR-based quantification of energetic phenotypes, including ATP-linked and spare respiratory capacities.
Cellular iron detection
Following cell lysis with RIPA buffer, intracellular iron levels were quantified using a commercial ferrozine assay kit (Leagene Biotechnology, Beijing, China). To quantify the labile iron pool (Fe2⁺), cells were stained with FerroOrange fluorescent probe (1 μM; Merck, Darmstadt, Germany). After PBS washing, cells were resuspended for flow cytometer (BD).
Fluorescence in situ hybridization (FISH)
The probe (labeled with Cy3) and FISH kit were purchased from RiboBio (Guangzhou, China). The cells were fixed in 4% polyformaldehyde for 20 min and permeabilized in 0.1% Triton X-100 for 15 min. After that, the cells were incubated with lncRNA-Gm5532 probe working solution at 37 °C in the dark overnight. Finally, the nuclei of the cells were stained with DAPI and photographed via a confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany).
RNA pulldown assay
RNA sense and antisense probes were transcribed in vitro from linear pUC19 cut by the Xba/restriction enzyme. Biotinylated RNAs were synthesized using T7 RNA polymerase according to the manufacturer’s protocol. RNA pull-down assays were conducted using the Magnetic RNA–Protein Pull-Down Kit (Pierce, Rockford, USA). The eluted proteins were detected with silver staining, mass spectrometry, and western blot.
Prediction of protein‒lncRNA interactions
The sequence of the iASPP protein was obtained from the UniProt website (https://www.uniprot.org/), and the protein crystal structure was simulated via I-TASSER (https://zhanggroup.org/I-TASSER/) and optimized via Rosetta's Relax module. 3dRNAs were used for RNA modeling (http://biophy.hust.edu.cn/new/3dRNAs) according to the Gm5532 sequence. HADDOCK was used for rigid docking. The output results were analyzed by the force analysis module in PDBePISA, and 3D conformation and force display were performed via PyMOL.
The RNA‒protein complex obtained via molecular docking was used as the initial structure for the full-atom molecular dynamics simulation, which was conducted with Gromacs 2020.6 software for 50 ns. The trajectories after simulation were periodically eliminated, and subsequent RMSD, RMSF, and secondary structure analyses were performed.
RPISeq (http://pridb.gdcb.iastate.edu/RPISeq/index.html) was used to predict the interaction probabilities of Gm5530 with iASPP. Predictions with a probability greater than 0.5 indicated potential interactions between RNAs and proteins.
The catRAPID algorithm (http://service.tartaglialab.com/page/catrapid_group) was used to estimate the interaction propensity and Gm5532 binding domains of iASPP by combining secondary structure, hydrogen bonding and van der Waals contributions (Armaos et al. 2021).
RNA immunoprecipitation
The RNA immunoprecipitation (RIP) assay was conducted using a RIP kit (Gene Create, Wuhan, China) according to the manufacturer’s protocols. Cells were lysed with a RIP lysis buffer. Magnetic beads (50 μL) mixed with iASPP antibodies (3 μg; Bioworld) or negative control IgG were incubated with the lysate (100 μL) overnight at 4 °C with rotation. The magnetic beads were digested with proteinase K at 55 °C for 30 min. RNA was purified by Trizol regent. The enrichment of Gm5532 was examined by qPCR method. The primers were listed in supplemental materials.
Cell transfection and transduction
The lentivirus overexpressing iASPP was constructed and purchased from GenePharma (Suzhou, China). A lentiviral empty vector was used as negative control (NC). The BMMs were seeded and cultured overnight, and were infected with lentivirus particles added at a multiplicity of infection (MOI) of 10. The transduced cells were screened with puromycin (1–5 μg/mL) one week later. si-iASPP, si-NRF2 and scrambled siRNA were synthesized by GenePharma. After the BMMs were seeded and cultured overnight, the siRNAs were transfected with siRNA mate (GenePharma). The scrambled siRNA was used as negative control (si-NC).
Co-IP
The cells were lysed in IP lysis buffer (Beyotime) supplemented with protease inhibitors. The lysate was precleaned with protein A/G magnetic beads (Beyotime) at 4 °C. Afterwards, the indicated antibodies or IgG were added and incubated on a rotating wheel at 4 °C overnight. Protein A/G magnetic beads were added and incubated on a rotating wheel at room temperature for two hours. The beads were boiled in SDS loading buffer and analyzed by Western blotting. The primary antibodies used for co-IP included iASPP (Bioworld, Nanjing, China), NRF2 (Beyotime), KEAP1 (Bioworld), and ubiquitin (Bioworld).
Chromatin immunoprecipitation
The chromatin immunoprecipitation (ChIP) assay was conducted via a ChIP kit (Gene Create, Wuhan, China) according to the manufacturer’s protocols. The cells were first lysed. The lysates were incubated with NRF2 (Bioworld, Nanjing, China) antibodies (3 μg) or negative control IgG overnight at 4 °C with rotation. After that, protein A/G magnetic beads were added to the lysate and incubated at 4 °C with rotation for 2 h. The magnetic beads were subsequently digested with proteinase K at 55 °C for 30 min. DNA was purified with Trizol regent. The enrichment of DNA fragments was examined via the qPCR method. The primers used were listed in supplemental materials.
Quantitative PCR
Cellular RNA was isolated and converted to cDNA through reverse transcription. mRNA expression was quantified employing the SYBR Green PCR Kit (Beyotime) under the conditions: 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 25 s. Gapdh functioned as the endogenous reference for mRNA quantification normalization. For assessment of mtDNA copy number, total cellular DNA was extracted. qPCR was conducted to quantify the mitochondrially encoded NADH dehydrogenase 1 (MT-ND1) and the constitutively expressed nuclear Gapdh. The mRNA expression and mtDNA copy number were analyzed using comparative Ct method. The custom-designed primer pairs were presented in supplemental materials.
Western blot
Cell lysis was performed using RIPA buffer (Beyotime), and equal aliquots (20 μg each sample) were separated by 10% SDS-PAGE and electrotransferred onto PVDF membranes. Following saturated in 5% skim milk, the PVDF membranes were probed with primary antibodies (dilution 1:1000) at 4 °C for 16 h, and then were incubated with HRP-labeled secondary antibodies (Beyotime), and target proteins were visualized employing ECL (enhanced chemiluminescence) reagents (Millipore, Billerica, MA, USA). The following primary antibodies were used: cathepsin K (CTSK; Beyotime), matrix metallopeptidase 9 (MMP-9; Beyotime), nuclear factor of activated T cells 1 (NFATc1; Abcam, Cambridge, UK), integrin beta 3 (ITGB3; Beyotime), p38 (Cell Signaling Technology, Danvers, MA, USA), p-p38 (Cell Signaling Technology), ERK (Cell Signaling Technology), p-ERK (Cell Signaling Technology), JNK (Abcam), p-JNK (Abcam), NDUFB8 (Bioworld), SDHB (Bioworld), UQCRC2 (Bioworld), MTCO1 (Bioworld), ATP5A (Bioworld), TfR1 (Beyotime), FTL (Cell Signaling Technology), FTH (Cell Signaling Technology), FPN1 (Abcam), iASPP (Bioworld), NRF2 (Beyotime), KEAP1 (Bioworld), ubiquitin (Bioworld) and GAPDH (Cell Signaling Technology). GAPDH functioned as the endogenous reference for protein quantification normalization.
Statistical analysis
Data were shown as mean ± SD. Comparative analysis across groups was executed as follows: two-group analysis, unpaired two-tailed t-tests; multi-group comparison, one-way ANOVA and Tukey's Post-Hoc test. All computations were conducted using Prism software (La Jolla, CA, USA) with P < 0.05 significance.

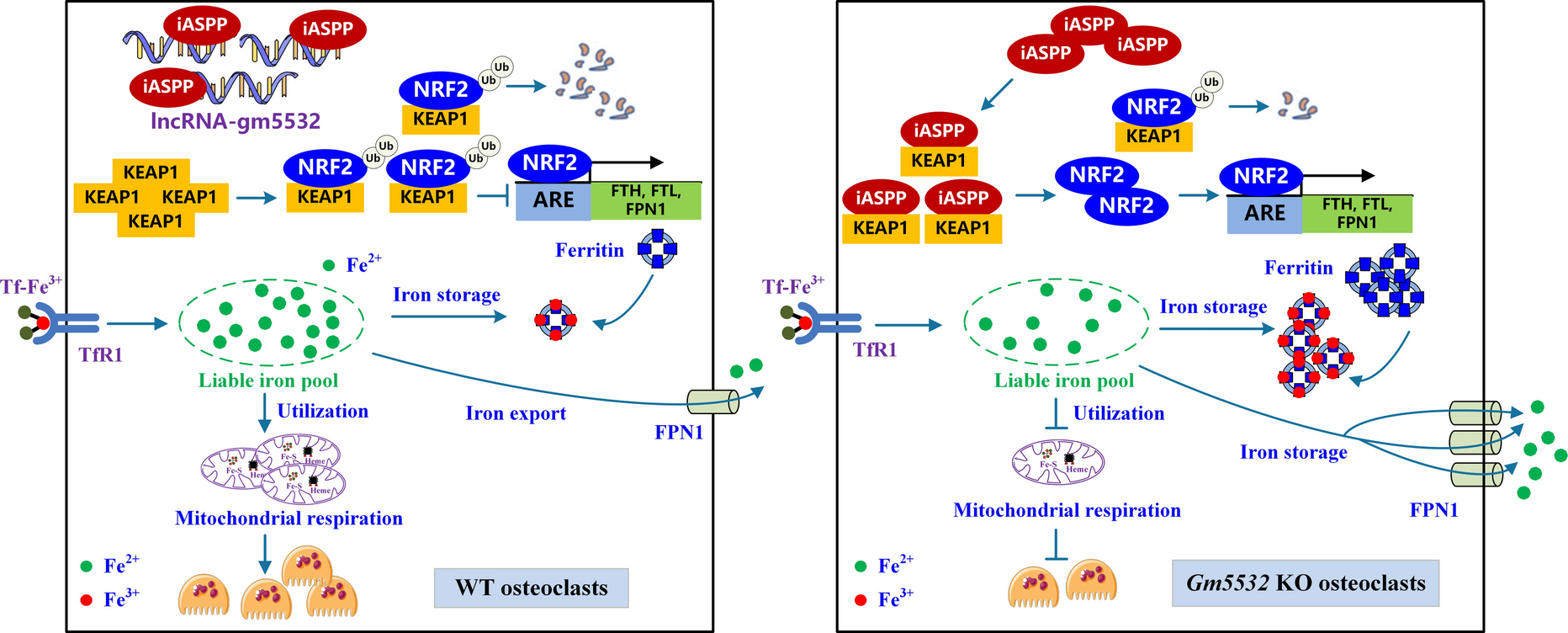
Comments (0)