Animals and ischemic model
The Guangzhou Huateng Biomedical Technology Co., Ltd. Animal Protection and Utilization Committee authorized this study (Ethic number: HTSW221143, Approval date: 19 April 2023). Adult male C57BL/6 mice (n = 120, 8–12 weeks old, 20–22 g; Guangzhou Huateng Biomedical Technology Co., Ltd., Guangzhou, China) were housed at 22 ± 1 °C, 12-h light/dark cycle, free access to food and water, with six animals/cage. The mice were assigned at random to sham-operated and model groups. Here, we followed the earlier published middle cerebral artery occlusion (MCAO) model to simulate transient cerebral ischemia (Zhang et al. 2018b, 2017, 2018c) by first subjecting the mice to 1 h of ischemia followed by reperfusion at 0, 6, 12, and 24 h before returning to their cages. The sham-operated group underwent the same surgical steps except for suturing and blocking, which induced MCAO. Unless otherwise stated, all experiments and assays followed the manufacturer's protocols.
Neurological deficits and cerebral infarct volume
Here, 24 h following MCAO/R, neurological deficits were validated by the modified Neurological Severity Score, which comprises four aspects: motor, sensory, reflex, and balance. The total score was 18 points; 0: normal neurological function in mice, 1–6: mild neurological injury, 7–12: moderate neurological injury, 13–18: severe neurological injury, and 18 points indicating a complete loss of neurological function (Chen et al. 2001). The mice were euthanized after neurological behavioral assessment. Using brain matrices, the brains were extracted and sliced into four 1 mm-thick slices. Brain infarctions were detected using 1.5% TTC staining (Hui et al. 2016).
In vivomodulation of METTL7B expression
Recombinant lentiviruses encoding METTL7B overexpression vectors, METTL7B-targeted short hairpin RNA vectors, and their respective negative controls were provided by PackGene (Guangzhou, China) to investigate the functional METTL7B role in vivo. Adult mice were anesthetized using isoflurane (1.5–3%) and positioned securely in a stereotactic apparatus. Following standard surgical protocols (Zeng et al. 2016), lentiviral solutions (3 μL at a viral titer of 2 × 109 TU/mL) were stereotactically infused into the right cortical area in the lateral ventricle at a controlled injection rate of 0.2 μL/min. The injection needle was maintained in place for another 3 min after administration to ensure effective viral distribution and minimize backflow. After a two-week recovery and expression stabilization period following lentiviral intervention, mice from each group were subjected to MCAO surgery to induce IS, enabling the assessment of the effects of METTL7B modulation on CIRI.
H&E staining and immunohistochemistry assays
Mouse brains were collected for histopathological evaluation 24 h after MCAO (n = 3 per group). Coronal sections were subjected to H&E and immunohistochemical analysis to assess infarct-induced tissue injury. Tissue fixation, staining, and microscopic assessment were conducted (Zhang et al. 2021).
TUNEL assay
Using the TUNEL assay, we evaluated neuronal apoptosis by first performing permeabilization with 0.1% Triton X-100 after fixing cultured cells or brain tissue slices in 4% paraformaldehyde at 4 °C for a minute. Following that, the samples were placed in a dark, humidified room and incubated with TUNEL reaction solution at 37 °C for an hour. Following labeling, DAPI was utilized as a counterstain for the nuclei. A fluorescent microscope (Leica DM4000, Wetzlar, Germany) was used to observe and image the cells that had undergone cell death. The ratio of TUNEL-positive cells to total cells was used to determine the apoptotic index.
Cell culture and oxygen–glucose deprivation (OGD) treatment
Seeking the assessment of the impact of in vitro IRI, we grew mouse neuroblastoma N2a cells (ATCC, CCL-131). The N2a Cells were grown in a medium that contained 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, besides DMEM. The N2a Cells were grown in anoxic and glucose-free DMEM with 5% CO2, 1% O2, and 94% N2 for 4 h in order to produce OGD. To replicate the effects of reperfusion damage, the cells were cultured in their original media for 12 h at 37 °C with 5% CO2.
Cell transfection
Neurons were exposed to transfection with lncRNA-MIR22HG and METTL7B siRNA (si-MIR22HG and si-METTL7B) or negative control siRNA to interfere with lncRNA-MIR22HG and METTL7B. The recombinant plasmid pcDNA3.1, containing the lncRNA-MIR22HG and METTL7B genes, was subjected to transfection into N2a Cells, with pcDNA3.1-NC empty plasmid as a control to overexpress lncRNA-MIR22HG and METTL7B. All transfections were performed using Lipofectamine 2000, and neuronal samples were collected for subsequent experimental studies after 48 h.
RNA fluorescence in situ hybridization (FISH)
To conduct FISH, we used lncRNA-MIR22HG-specific fluorescence-conjugated probes. Following OGD/R exposure, neuronal cells were cultured in a non-denaturing environment and hybridized with lncRNA-MIR22HG probes. The N2a Cells were counterstained with DAPI for 10 min following hybridization-induced cell death. A confocal microscope (LSM10, Zeiss, Oberkochen, Germany) was utilized to probe the fluorescent signals.
Cell viability measurement
The viability of N2a Cells subjected to OGD following lncRNA-MIR22HG and METTL7B knockdown or overexpression was quantitatively ascertained with CCK-8 (Sigma-Aldrich, St. Louis, MO, USA). The N2a Cells were plated (4 × 104 cells/cm2) in 96-well plates, and each well was supplemented with 10 μL CCK-8 reagent (5 mg/mL) at the end of the experimental OGD treatment and incubated at 37 °C for an hour. The reaction medium was carefully aspirated and replaced with 150 μL DMSO/well to solubilize formazan crystals. Optical density was determined spectrophotometrically at 450 nm by a microplate reader. Viability was reported as a percentage relative to control N2a Cells cultured under normal conditions without exposure to OGD/R.
Lactate dehydrogenase assays
The lactate dehydrogenase (LDH) concentration released into the culture supernatant was quantified to assess OGD/R in N2a Cell-provoked cytotoxicity (Zhong et al. 2019).
Annexin V-FITC/propidium iodide (PI) staining
Neurons seeded in 12-well culture plates were exposed to OGD for 4 h, then reoxygenated for 24 h to quantitatively evaluate neuronal apoptosis. The cells were harvested and rinsed three times with PBS. Harvested N2a Cells were stained at ambient temperature for 25 min in darkness using a mixture comprising 6 μL of annexin V-FITC and 12 μL of PI. The stained neuronal populations were immediately subjected to flow cytometry. Apoptosis rates were evaluated relying upon the cumulative proportion of N2a Cells identified in the early apoptotic phase (annexin V-positive and PI-negative) and those classified as late-stage apoptotic (annexin V-positive and PI-positive).
Methylated RNA immunoprecipitation (IP) assay
The solutions were well mixed by gently shaking or tilting the plate to provide comprehensive coverage of each well for methylation RNA IP. To create a standard curve, single-point controls were generated by adding 2 μL of positive control RNA (0.5 ng/μL) to each well, resulting in final concentrations of 0.02, 0.1, 0.2, 0.4, and 1 ng/well. To ensure optimal antibody binding effectiveness, the total RNA intake for each sample was limited to a maximum of 8 μL. The samples were meticulously observed throughout incubation to detect any colorimetric changes. Subsequently, 100 μL of blocking buffer was introduced to each well to suppress non-specific enzyme activity. The reaction was concluded by adding a stop solution, leading to a yellow color shift. Absorbance was quantified at 450 nm utilizing a microplate reader within 2 to 10 min following the stop solution addition.
RNA stability assessment
N2a cells at confluence received actinomycin D (5 μg/mL; Sigma-Aldrich) to inhibit transcription and evaluate RNA stability. Cells were harvested at 0, 1, 2, 4, and 6 h after adding actinomycin D. At each time point, the cells were washed twice with ice-cold PBS and lysed using TRIzol reagent for total RNA extraction. Subsequently, 1 mg of purified RNA was reverse-transcribed using gene-specific primers. The lncRNA-MIR22HG and housekeeping GAPDH gene transcript levels were determined via qRT-PCR using an ABI 7900HT system. Relative mRNA abundance was determined via the 2–ΔCt methodology (ΔCt = Ct_target – Ct_GAPDH) and normalized to baseline (0 h). RNA decay rates (first-order degradation constants, k) and half-lives (t₁/₂ = ln2/k) were calculated from linear regression analysis of the natural logarithm of relative transcript levels versus time. Each experiment was conducted in triplicate, and the procedure was independently replicated thrice.
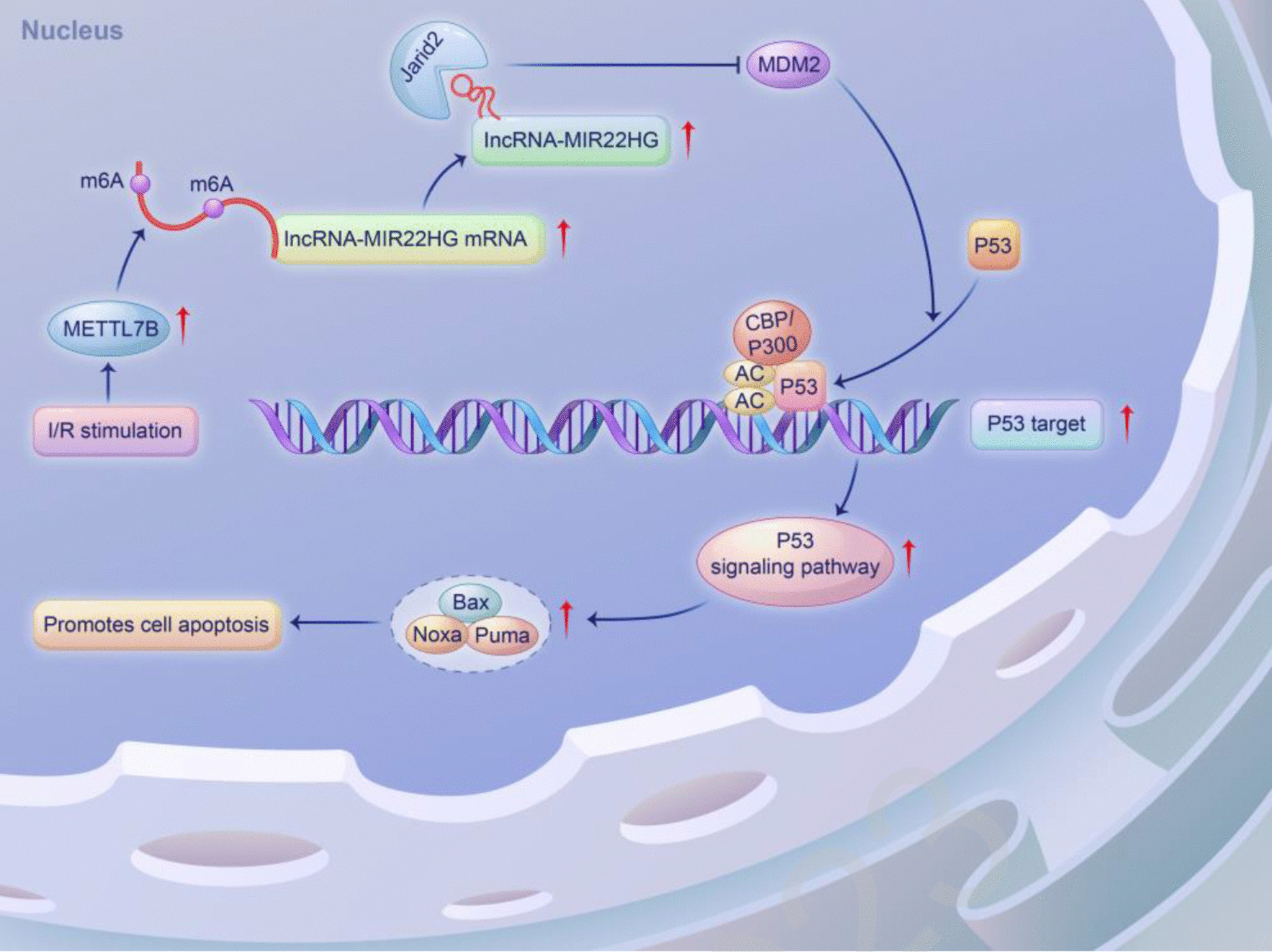
RNA IP and RNA pull-down assay
N2a cells underwent OGD/R with a control group created under normal conditions. Subsequent to OGD/R, the initial culture media were eliminated, and the cellular precipitates were collected. Cell lysate samples were allocated into three primary fractions: 0.8 mL for IP, 0.8 mL for immunoglobulin G (IgG), and 0.1 mL for input. RNA samples (IP, IgG, beads, and input) underwent reverse transcription. A designated quantity of the PCR result was thereafter subjected to electrophoresis on a 3% agarose gel to confirm amplification efficacy. Concurrently, RT-PCR was performed for data analysis, demonstrating the interaction between lncRNA-MIR22HG and JARID2. The N2a cells were lysed using the cell lysate from the RNA pull-down test by adding 1.7 mL of RIP buffer, 17 μL of protease inhibitor cocktail, and 17 μL of DTT, followed by homogenization 15–20 times. The mixture was subsequently centrifuged (5,000 g, 15 min, 4 °C), and the supernatant was moved to a fresh centrifuge tube. The RPD and NC samples were isolated and rinsed, magnetic beads were retrieved using a magnetic rack, and the supernatant was moved to a fresh centrifuge tube. Following SDS-PAGE and silver staining, the protein samples were examined using mass spectrometry for protein identification.
Chromatin IP assay
Neuronal fusion was allowed to reach approximately 80%, after which the cells were transfected, and the experiment was initiated 36 h later. The cells were then fixed and incubated at room temperature for 10 min. Thereafter, 1 mL of 2.5 M glycine was introduced, and the cells were re-incubated in the same conditions to terminate the fixation reaction. The cells were washed 2–3 times with ice-cold EDPC-PBS, scraped, and collected in a centrifuge tube. The cells were centrifuged (3000 rpm/min, low temperature, 3 min) to obtain the cell pellet. The cells were then resuspended by pipetting, and the suspension was transferred to a 1.5 mL centrifuge tube (4 °C, 2000 rpm, 5 min). Cell lysis buffer containing phenylmethylsulfonyl fluoride and cocktail was introduced to resuspend the cells, which were placed on ice for 10–20 min. The cells were centrifuged (4 °C, 3000 rpm, 5 min), the supernatant was discarded to obtain the cell nuclei, and 1 mL of RIPA lysis buffer containing phenylmethylsulfonyl fluoride and cocktail was added to resuspend the cells. They were placed on ice for 10–20 min and incubated on a shaker at 4 °C for 10 min. The DNA was sheared to sizes between 200 and 1000 bp in an ice-water bath using ultrasonication. The shearing conditions were 30% power, 30 s on, and 30 s off, totaling 30 pulses. The ultrasonication probe was kept below the liquid surface to avoid bubble formation.
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA extraction from neuronal cultures and C57BL/6 mouse brain specimens was carried out utilizing TRIzol reagent (Invitrogen, Waltham, MA, USA) and reverse transcribed with 1 μg of RNA utilizing gene-specific primers and commercial cDNA synthesis kits (Thermo Fisher Scientific, Waltham, MA, USA) for mRNA and miRNA targets. The qRT-PCR analysis of lncRNA-MIR22HG, miR-29b, and HMGB1 transcripts was conducted utilizing the ABI Prism 7900HT equipment (Applied Biosystems, Waltham, MA, USA) in conjunction with All-in-One qPCR Mix (GeneCopoeia, Rockville, MD, USA) as follows: initial denaturation at 94 °C for a specified duration, 40 cycles at 94 °C for 1 min, 60 °C for 30 s, and 72 °C for 1 min, culminating in a final extension at 72 °C for a specified duration. All reactions were conducted in triplicate to guarantee repeatability. The 2−ΔΔCt approach was utilized for relative quantification, employing GAPDH or U6 small nuclear RNA as internal normalization controls. All reactions were conducted in triplicate to guarantee repeatability. Table 1 provides the primer sequences.
Table 1 Primers of qRT-PCRWestern blot (WB) analysis
Total protein was extracted from N2a cells and mouse brain tissue by homogenization in ice-cold RIPA buffer (Haimen Biotechnology Co., Jiangsu, China) for 30 min, determining protein concentrations using the BCA assay. Equal protein amounts (≤ 50 μg per lane) were resolved on 12% SDS-PAGE (Sigma-Aldrich) and electrophoretically transferred onto nitrocellulose membranes (Pall Corporation, Port Washington, NY, USA) blocked for 2 h at room temperature in 5% nonfat dry milk, followed by overnight incubation at 4 °C with the following primary antibodies: MDM2 (Affinity, AF6376, 1:1000), p300 (Affinity, A3752, 1:1000), Bax (Servicebio, GB114122, 1:1000), Puma (Abclonal, A3752, 1:1000), Noxa (Affinity, DF13193, 1:1000), JARID2 (Abcam, ab308119, 1:1000), METTL7B (Abclonal, A7200, 1:1000), p53 (Servicebio, GB11626, 1:1000), and β-actin (Servicebio, GB15004, 1:10,000). After rinsing with Tris-buffered saline containing 0.1% Tween-20, membranes were incubated for 1 h at room temperature with horseradish peroxidase–conjugated goat anti-rabbit secondary antibody (Servicebio, GB15003, 1:5000) targeting β-actin. Immunoreactive bands visualization was applied by an ECL detection kit (Pierce Biotechnology, Waltham, MA, USA), and band intensities quantification by Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA). All experiments were performed in triplicate.
Statistical analyses
Data are reported as the mean ± SD. Statistical analysis was carried out via one-way ANOVA, followed by Tukey's post-hoc test for multiple comparisons using SPSS version 27.0. Each experiment was independently repeated at least three times. P < 0.05 was deemed statistically significant.

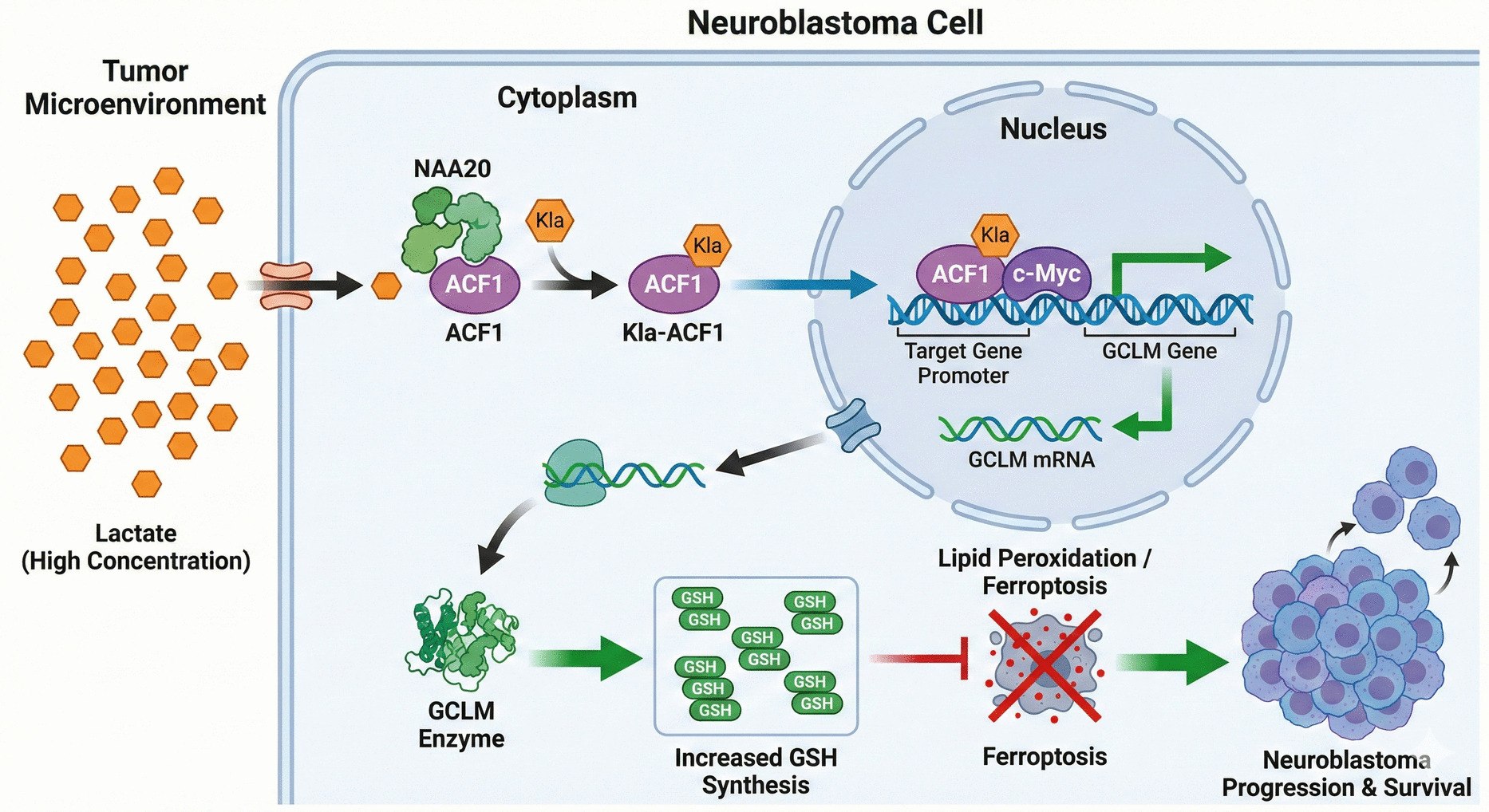
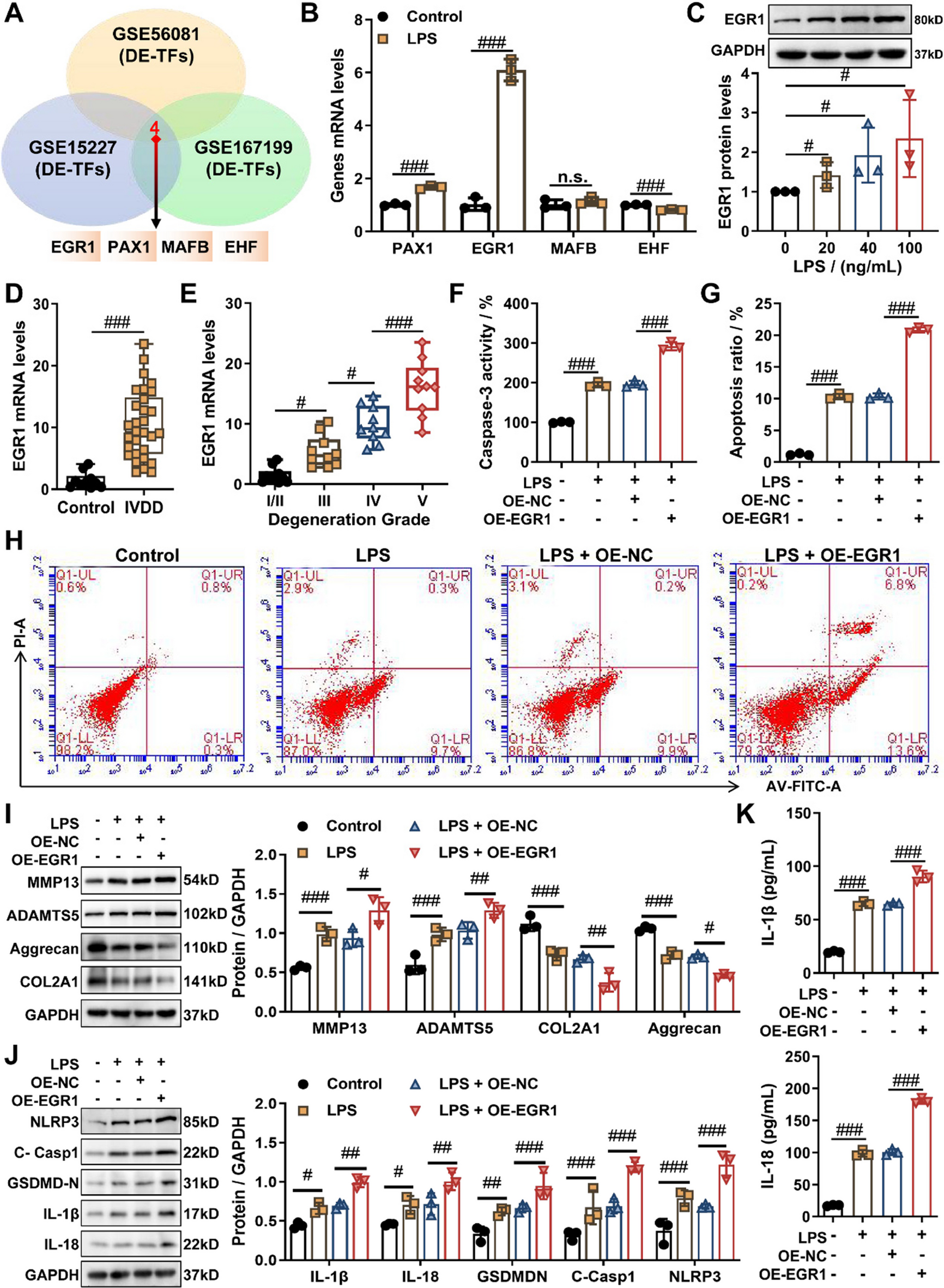
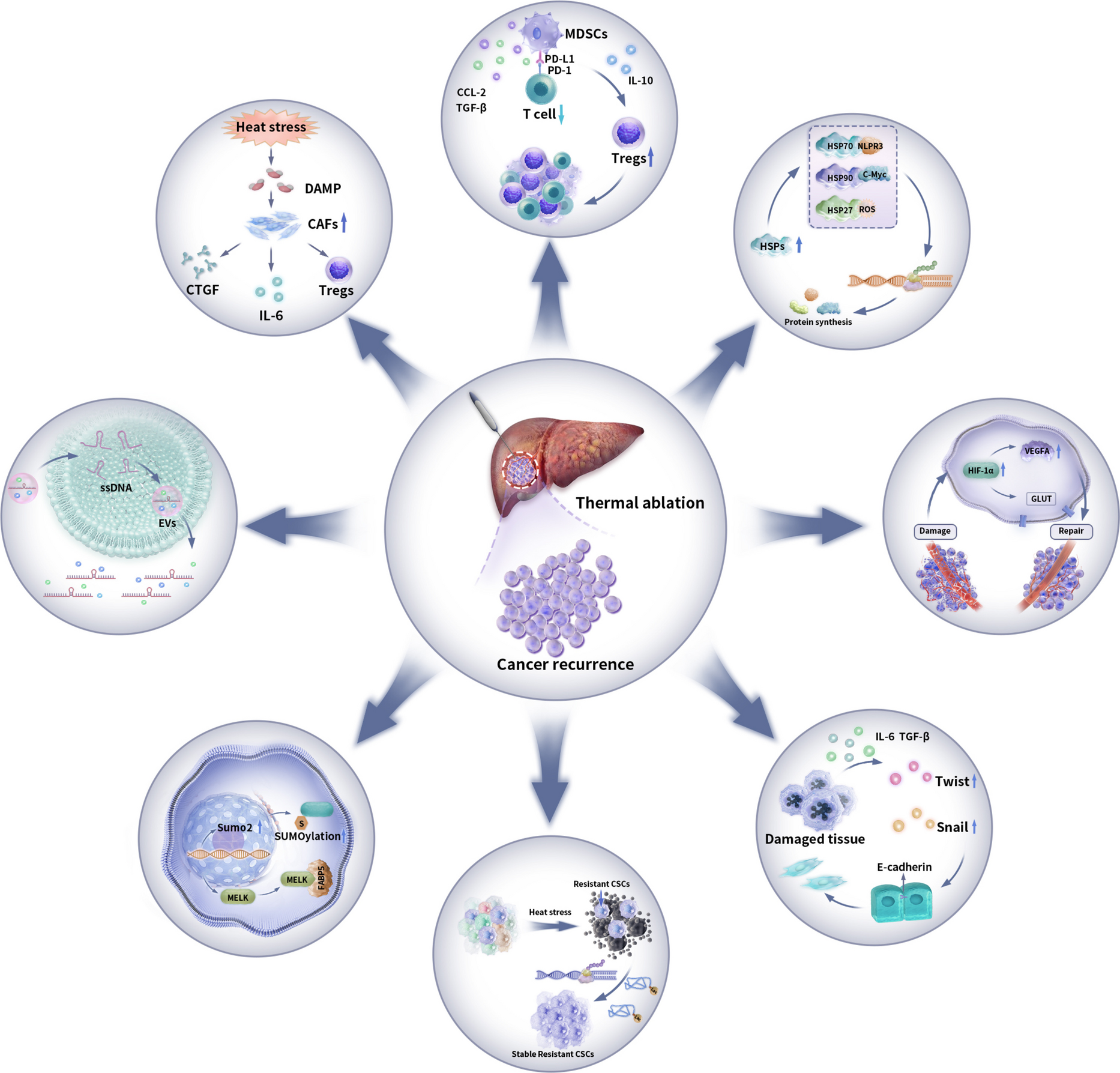
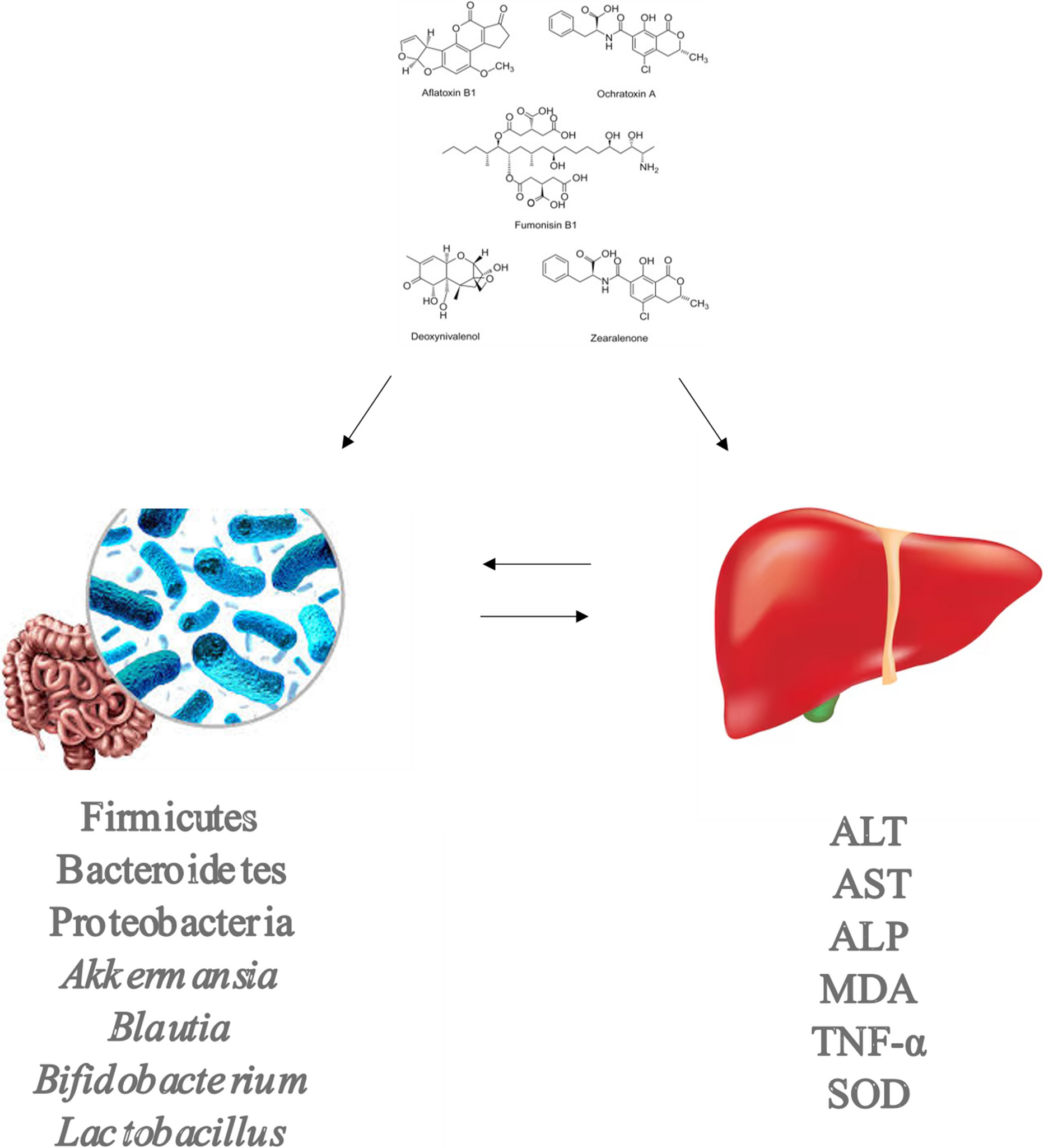
Comments (0)