Bacterial growth condition
The plant pathogen R. solanacearum (GenBank: KC888020), originally isolated from Longgang Town, Kaiyang County, Guiyang City, Guizhou Province (107.0969 E, 26.8819 N), was used as the model pathogen in this study. The pathogen R. solanacearum grew in NB medium (nutrient broth, glucose 10 g/L, beef extract powder 3 g/L, peptone 5 g/L, yeast extract 0.5 g/L, with agar powder 16 g/L for solid medium, adjusted pH 7.0 ~ 7.2). The R. solanacearum cultures were washed twice with sterile 0.9% NaCl, and the OD600 was adjusted to 0.6 (equivalent to approximately 108 colony-forming units per milliliter, CFU/mL) before all experiments. Isolated strains were grown in TSB medium (tryptic soy broth, tryptone 15 g/L, soy peptone 5 g/L and NaCl 5 g/L, and agar powder 16 g/L for solid medium).
Extraction of tobacco root exudate
The tobacco root exudate extraction method was modified from Liu et al. (2015). Tobacco cultivar Yunyan87 (Nicotiana tabacum L.) was used as the model plant. Tobacco seeds were soaked in sterilized deionized water three times to prepare for germination. Seedlings were grown using the floating seedling method for 60 days. Healthy tobacco seedlings were selected, and the root-attached substrate was gently rinsed off with sterile ultrapure water, followed by three additional rinses. The seedlings were then fixed in a planting box with sterile sponges, with a light/darkness 12/12 h, and a temperature of 25 °C. During the cultivation phase, Hoagland nutrient solution and micronutrient solution (Maurer et al. 2021) were continuously added to the water to support the normal growth of the seedlings. An electromagnetic pump (RESUN, Shenzhen, China) was used to oxygenate the nutrient solution. Ten tobacco plants with same size and well-developed root system were selected, the roots were washed by sterilized water, and were put into a 250-mL sterilized water beaker wrapped in black cloth. The beaker then was placed in a climate chamber (light/darkness: 12 h/12 h, 25 °C) for 72 h. The root exudate solution was filtered through a microporous filter membrane made of organic polyvinylidene fluoride to remove impurities, immediately lyophilized, concentrated, and stored at − 20 °C for metabolites detection.
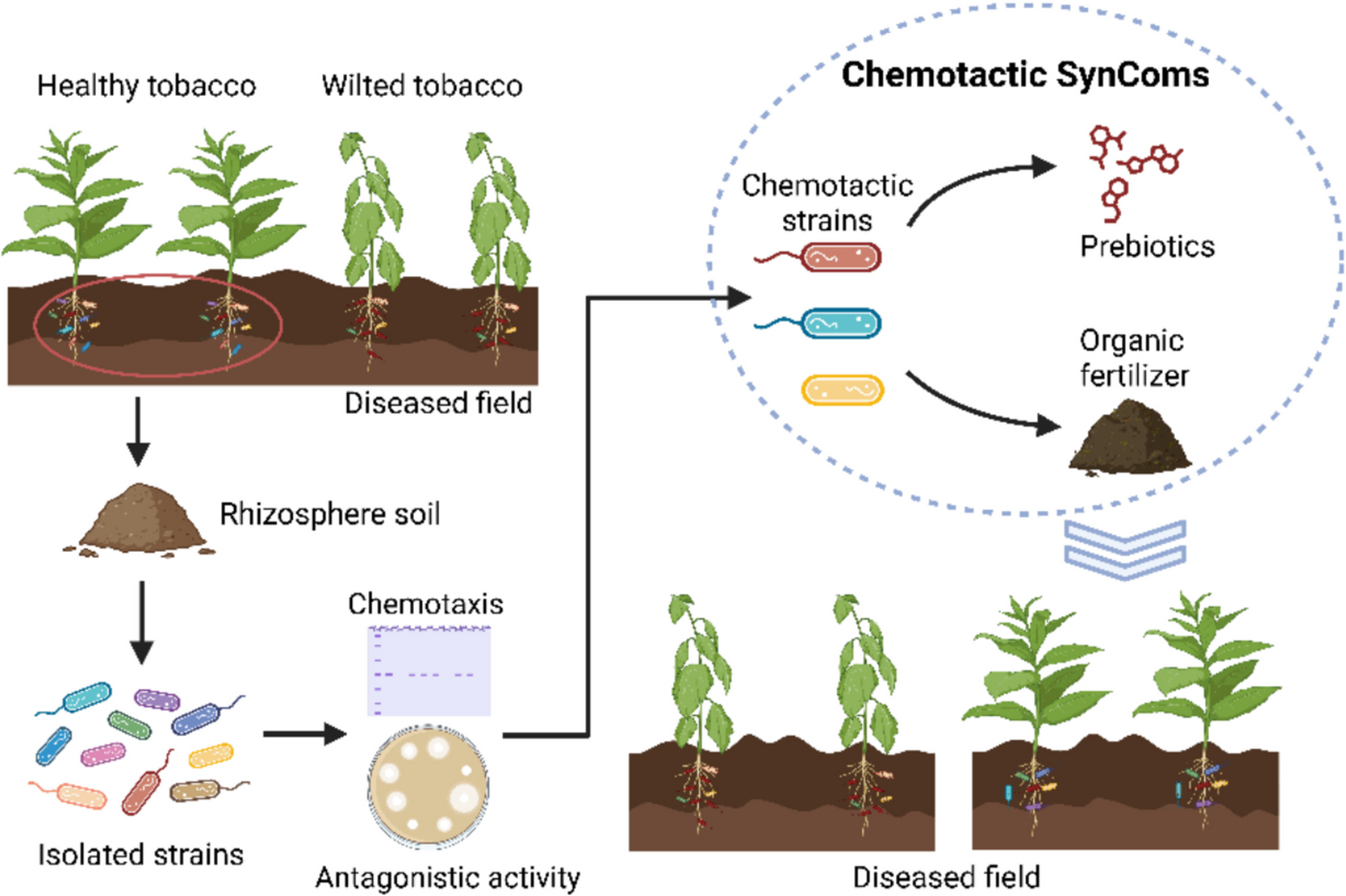
High-throughput isolation of bacteria from rhizosphere soil
The high-throughput isolation method of bacteria from soil was modified by Zhang et al. (2021). For greenhouse experiments, soil was collected from the Fuquan Town, Guizhou Province, China (107.5132°E, 26.7020°N), which has a long history of tobacco cultivation (pH: 4.4, organic matter: 33.51 g/kg, total nitrogen: 1.85 g/kg, total phosphorus: 1.70 g/kg, total potassium: 8.03 g/kg).
Tobacco plants were grown in pots under controlled greenhouse conditions (22 °C, > 85% relative humidity, 12 h light/12 h dark photoperiod). Six-week-old healthy tobacco plants were carefully uprooted, and loosely attached soil was gently shaken off. The remaining soil tightly adhering to the roots was collected as rhizosphere soil, and 1 g of rhizosphere soil was suspended in 9 mL of PBS buffer (phosphate-buffered saline). The homogenous solutions were transferred into 50-mL Falcon tubes and added to 25 mL MgCl2 solution for 15 min at room temperature. A 0.5 mL homogenous solution was added into 250 mL of root exudate extracted from the previous method, transferred into a 96-well plate, and incubated at 30 °C and 100 rpm for 48 h. The turbid cultures were stroken onto 50% TSB medium with agar to isolate strains. Each sample solution was repeated for three times. Strains with different colony morphologies were selected for subsequent experiment.
Assembly of chemotactic SynCom with antagonism against R. solanacearum
To generate a SynCom using isolated strains, we assessed the antagonistic activity of these strains against R. solanacearum using a spraying plate confrontation method, as described by Li et al. (2017). Sterilized water was used as control. The diameters of inhibition zones were measured. Then, the compatibility between strains was checked using the cross-streak method.
In order to determine whether the antagonistic bacteria possess chemotaxis, we used the histidine kinase gene cheA as a molecular marker. According to Buchan’s method (Buchan et al. 2010), a pair of primers, P4P5-F (5′-GGIMGIGGIGTIGGIATGGAYGTIGT-3′) and P4P5-R (5′-CCRTCICCIARIATIGTIGCICC-3′), was used for PCR. Based on a heuristic algorithm (Karkaria et al. 2021), we designed a SynCom with three strains possessing chemotaxis and antagonism against R. solanacearum. Briefly, this approach involved ranking all candidate strains based on their antagonistic activity against R. solanacearum. Strains with the highest antagonistic potential were prioritized for inclusion in the SynCom. If incompatibility was detected among selected strains, substitutions were made iteratively until a compatible combination was achieved.
Identification of targeted strains
The 16S rDNA of three strains described above was amplified by colony PCR (polymerase chain reaction) using general bacterial primers for 16S rDNA (forward primer 27F: 5′-AGAGTTTGATCCTGGCTCAG −3′, reverse primer 1492R: 5′-GGTTACCTTGTTACGACTT-3′). PCR products were sent to Sangon Biotech (Shanghai, China) for Sanger sequencing. The sequences obtained were aligned against the NCBI 16S rRNA database by BLAST. The top-matched reference strains, along with the sequenced strains, were used to construct phylogenetic trees via iTOL (https://itol.embl.de/).
Resource utilization patterns of the chemotactic bacterial isolates and R. solanacearum
To assess resource utilization of chemotactic bacterial isolates, 60 distinct resources commonly found in tomato root exudates, including amino acids, organic acids, and sugars, were purchased individually and tested (Supplemental Table S1). Chemotactic bacterial strains were first grown overnight in TSB medium at 30 °C with shaking at 150 rpm. Cells were collected, washed three times with 0.85% (w/v) NaCl, and resuspended to an OD₆₀₀ of 0.5. For each treatment, 10 μL of the bacterial suspension was added to a 96-well microtiter plate containing 150 μL of 1/10 TSB medium supplemented with 10 mM of a single resource. Plates were incubated at 30 °C with shaking at 170 rpm for 48 h, and bacterial growth was measured using a SpectraMax M5 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
For R. solanacearum, it was grown overnight in NB medium at 30 °C with shaking at 150 rpm. Cells were collected, washed three times with 0.85% (w/v) NaCl, and resuspended to an OD₆₀₀ of 0.5. For each treatment, 10 μL of the R. solanacearum suspension was added to a 96-well microtiter plate containing 180 μL of 1/10 NB medium supplemented with 10 mM of a specific resource. Plates were incubated at 30 °C with shaking at 170 rpm for 24 h, and bacterial growth was measured using a SpectraMax M5 spectrophotometer.
Effects of prebiotic-treated bacterial fermentation broth on the growth of R. solanacearum
Chemotactic bacterial cultures were grown to an OD₆₀₀ of 0.5 and inoculated into TSB medium containing 20 μL of a 1 mM solution of the selected resource in a 96-well microtiter plate. Mock treatment contained sterile water instead of the resource. Plates were incubated at 30 °C with shaking at 170 rpm for 48 h. Following incubation, cultures were centrifuged at 2000 rpm for 15 min. Residual bacterial cells were removed by centrifugation followed by filtration through a 0.22-μm membrane, and the supernatant was collected. Subsequently, 2 μL of a R. solanacearum suspension (OD₆₀₀ = 0.5) was added to NB medium supplemented with filter-sterilized supernatants. Cultures were incubated at 30 °C, 170 rpm for 48 h, and growth of R. solanacearum was assessed by measuring OD₆₀₀.
Creation of bacterial fertilizer for chemotactic SynCom
The bacterial strains were activated in TSB medium with agar at 30 °C for 12 h. We used an inoculation loop, transferred a single colony into 10-mL TSB culture medium, and placed it in a shaker at 30 °C and 170 rpm for 16 h. The fermentation broth was centrifuged at 5000 g for 10 min to collect bacterial cells.
We formulated five composite organic fertilizer groups using different mass ratios of four commonly used organic materials in tobacco cultivation, including vermicompost, distiller’s grains, cow dung, and oil cake, named S1 to S5 formulations (Table 1). Each strain used in the chemotactic SynCom was inoculated into the composite nutrient at a 5% (v/w) ratio, along with the corresponding prebiotics screened in the previous experiment. The moisture content was adjusted to 40%, and fermentation was carried out at 23 °C. We set up three replicates and prepared a bioorganic fertilizer using a two-stage solid-state fermentation method. During fermentation, the mixture was turned every 12 h, and a moisture content was maintained at 40% to ensure aeration and looseness of the compost. The fermentation period was approximately 7–10 days.
Table 1 Organic fertilizer formulations in this studyThe method for testing the fermentation capacity of the organic materials was based on the patent of Liu et al. (2018). The bioorganic fertilizer was weighed after fermentation in different proportions and diluted in sterilized NaCl solution (0.85%). The absorbance of each well was recorded after 48 h in a 96-well microplate for cultivation and analysis.
Soil microbial DNA extraction and 16S rRNA genes amplicon sequencing
Rhizosphere soil samples were collected from three healthy plants in the field treated with chemotactic SynCom, whereas three samples from a diseased field served as control. Genomic DNA was extracted using the PowerSoil® DNA Isolation Kit (MoBio, Carlsbad, CA, USA), following the manufacturer’s instructions. The extracted DNA was quantified using a Qubit 3.0 (Invitrogen, Waltham, MA, USA). A two-step dual indexing strategy was used for Illumina MiSeq (Illumina, San Diego, CA, USA). Bacterial community composition was determined by amplification of the V4-V5 region of the 16S rRNA gene with primers 563 F (forward primer: 5′-AYTGGGYDTAAAGVG-3′) and 802R (reverse primer: 5′-TACNVGGGTATCTAATCC-3′). The PCR products were purified using an AxyPrep PCR Clean-up Kit (Axygen Biosciences, Union City, CA, USA) before performing agarose gel electrophoresis. The concentrations of the purified amplicon products were determined using QuantiFluor-ST (Promega, Madison, WI, USA) and sequenced on an Illumina Hiseq 2500 platform at Shanghai Biozeron Biological Technology (Shanghai, China).
Plant treatments in the field
We selected a wilt-diseased field in Fuquan town, Guizhou Province as the experimental field. The treatment and control groups were set in the same field plot. A mixture of 100-g bacterial fertilizer and corresponding prebiotics (mass ratio of 99:1) was applied to a hole 15 cm deep and located 10 cm from the base of the tobacco plant. The control treatment followed conventional fertilization practices, including the application of 100 g of cow manure per plant.
Disease index was calculated using the formula described by Liu et al. (2013). Briefly, these plants were categorized into discrete severity levels based on visible symptoms: level 0 (no symptoms), level 1 (mild wilting, < 25% affected), level 2 (moderate wilting, 25–50% affected), level 3 (severe wilting, 51–75% affected), and level 4 (very severe wilting or plant death, > 75% affected). Each level was assigned a corresponding numerical value (0 to 4), and the disease index was calculated using the following formula:
$$Disease\;index=100\times\frac^n(n_i\times v_i)}}$$
where
\(\):
number of plants in severity level,
\(\):
disease severity score assigned to level,
N:
total number of surveyed plants,
\(v_\):
the maximum value of the disease severity scale
Agronomic traits, including leaf number, height of tobacco plants, stem perimeter, and leaf area index, were recorded before harvest.
Quantitative PCR analysis
Quantitative PCR (qPCR) was performed using primers for detection. The fliC gene encoding the flagella subunit (forward primer: 5′-GAACGCCAACGGTGCGAACT-3′; reverse primer: 5′-GGCGGCCTTCAGGGAGGTC-3′), was the specific gene for R. solanacearum quantification (Schonfeld et al. 2003). PCR was performed using the SYBR Premix Ex Taq™ kit (Takara, Kusatsu, Shiga, Japan). The PCR system (20 µL) consisted of 10 µL SYBR Premix Ex Taq, 0.4 µL ROX Reference Dye, 0.4 µL each of primer (5 µmol/L), 2 µL DNA template, and 6.8 µL ddH2O. The PCR conditions were as follows: 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, for 30 cycles.
Data analysis
All data analyses were performed using R version 4.3.1. Microbiome community diversity was calculated as ASV (amplicon sequence variant). LorMe package (https://cran.r-project.org/web/packages/LorMe) was used for microbial analyses. In detail, the alpha diversity was calculated via the “vegan” package (Dixon 2003). Principal coordinate analysis (PCoA) was conducted via the package “ape” (Paradis et al. 2004) based on the Bray‒Curtis distance matrix. Functional prediction of the microbial communities was performed using Tax4Fun (Aßhauer et al. 2015). The ComplexHeatmap package was utilized to depict the heatmap of bacterial resource utilization ability (Gu et al. 2016).

Comments (0)