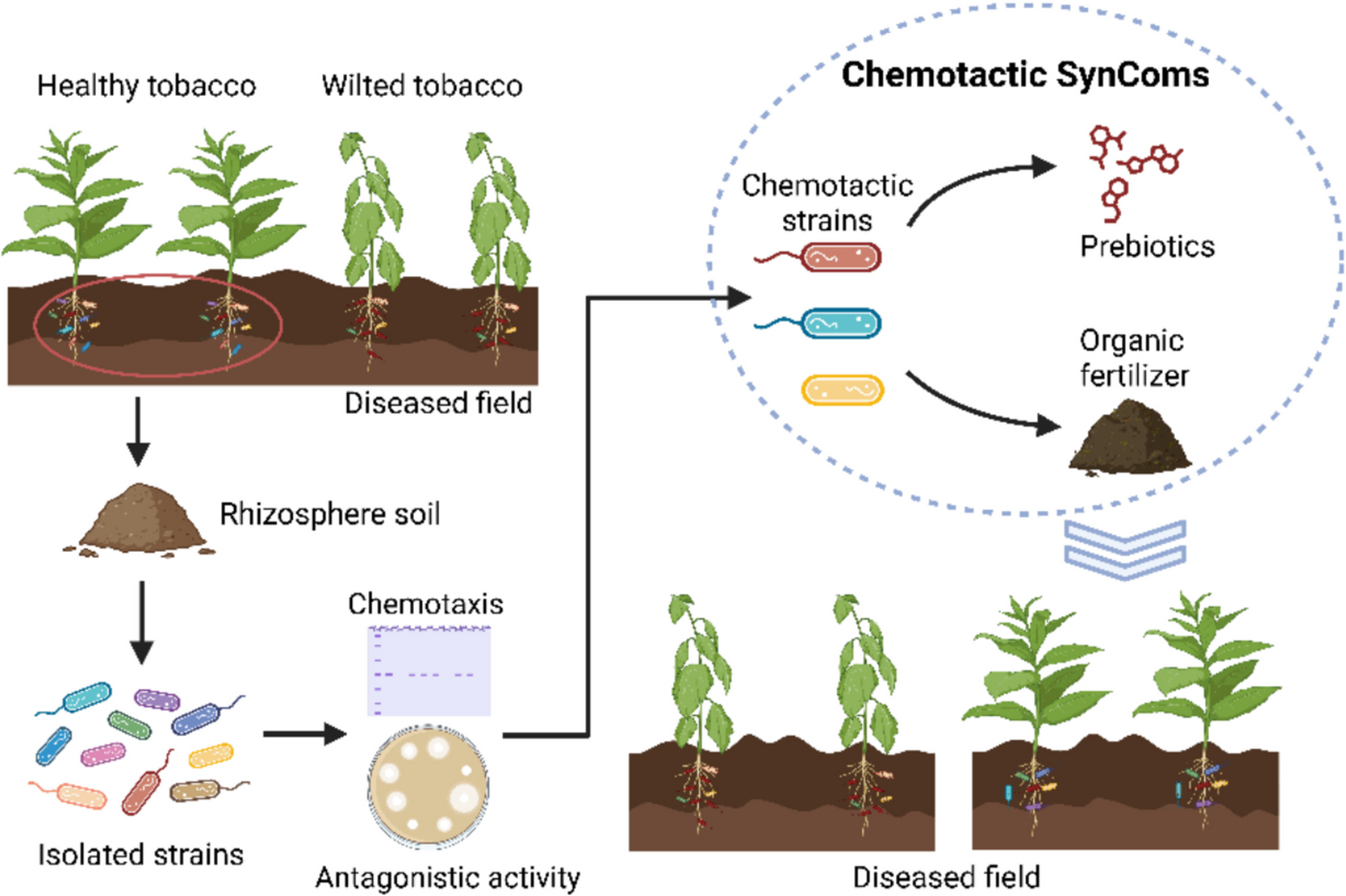
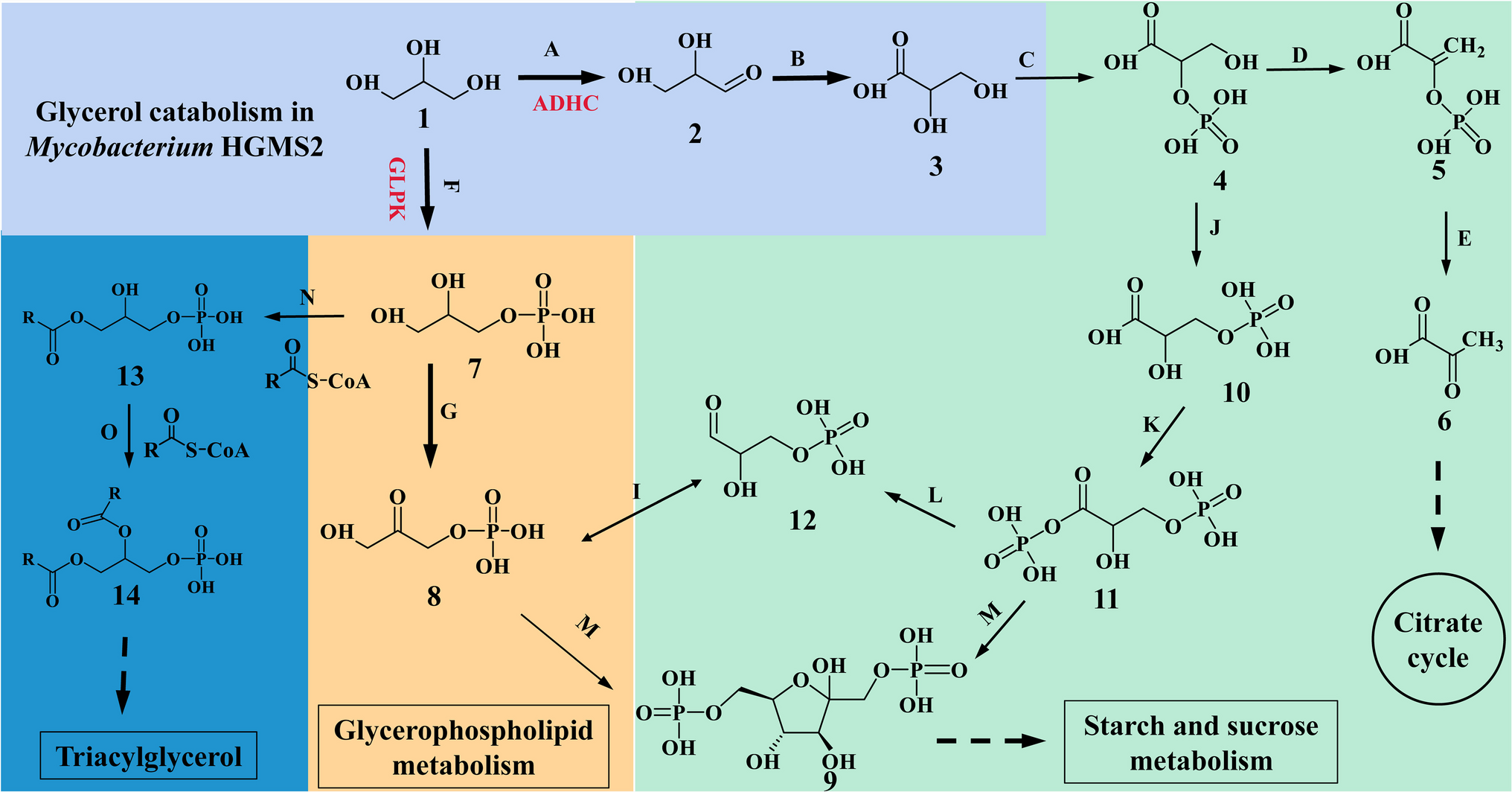
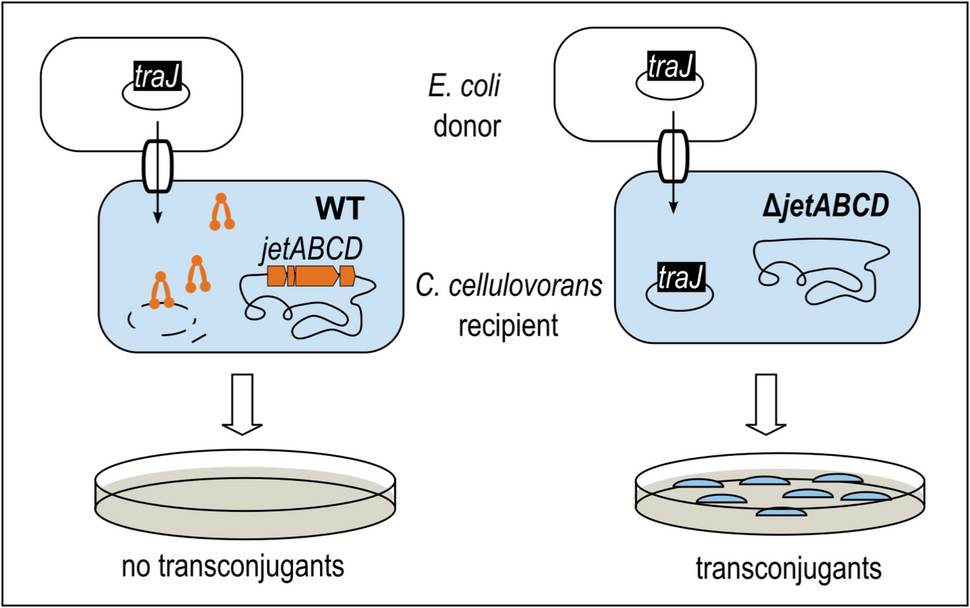
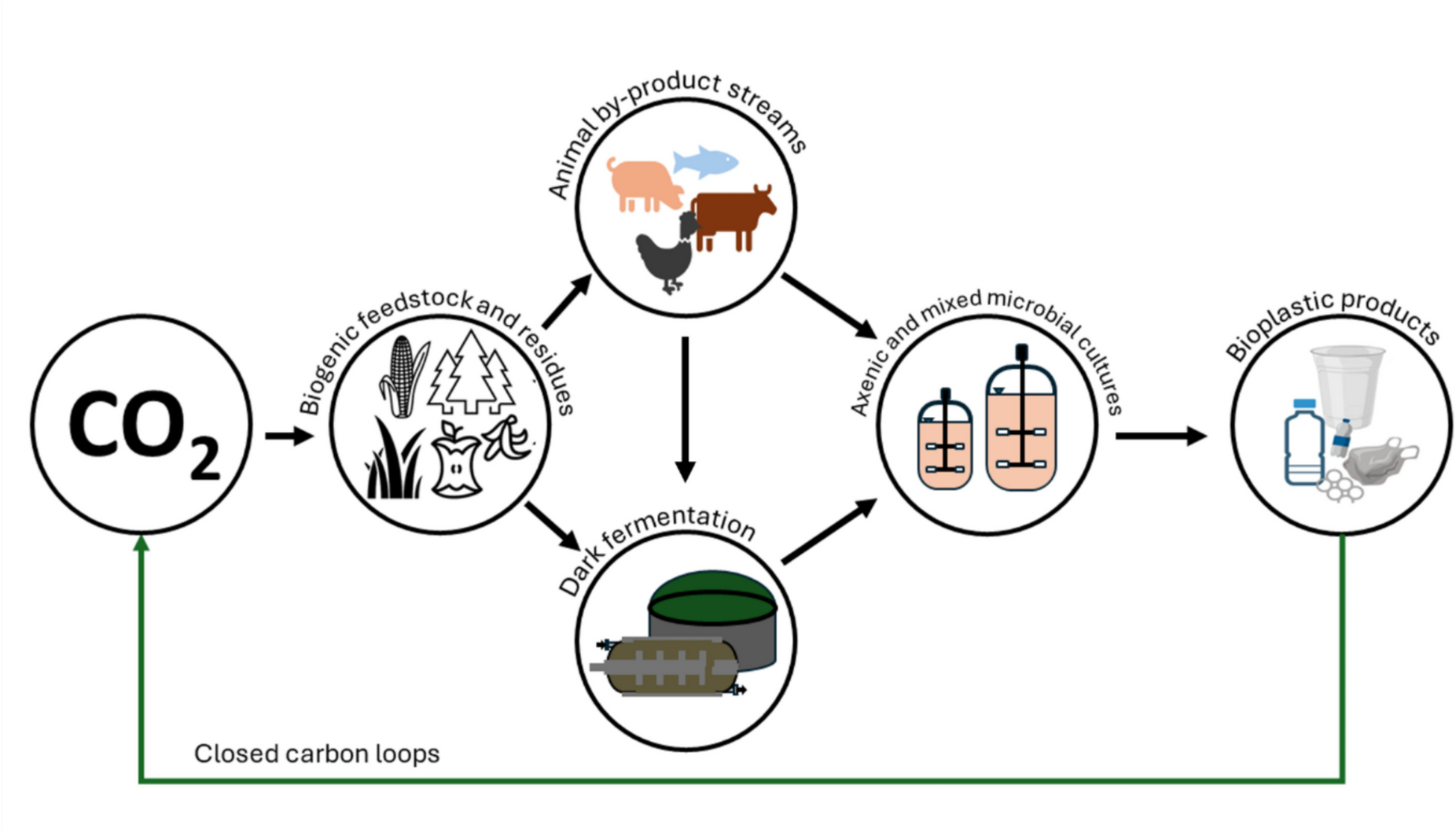
All chemicals and reagents used in this study were of laboratory grade and purchased from Sigma-Aldrich/Merck (St. Louis, MO, USA) unless otherwise stated.
Plasmid isolation
Plasmids pRS42-G-ChX, pRSCG_11, pSED1p-DIT1t, pTDH3p-DIT1t, pRDH182, and pRDH177_SED1 (Table 1) were propagated in Escherichia coli DH5α (Thermo Fisher Scientific, Waltham, MA, USA). These strains were streaked out from 40% (v/v) glycerol stocks stored at − 80 °C onto Luria–Bertani (LB) agar (5 g/L yeast extract, 10 g/L tryptone, 10 g/L NaCl, and 20 g/L agar) supplemented with 100 µg/mL ampicillin and incubated at 37 °C overnight. To prepare for plasmid extraction, resulting colonies were inoculated in liquid LB supplemented with 100 µg/mL ampicillin and incubated at 37 °C on a rotary wheel overnight. Plasmid DNA was extracted using a Zyppy Plasmid Miniprep Kit (Zymo Research, Irvine, CA, USA) as directed by the manufacturer.
Table 1 Plasmids used in this studyPlasmid construction
The plasmids pSED1p-DIT1t and pTDH3p-DIT1t were ordered as synthetic constructs from GeneArt (Regensburg, Germany). The plasmids contain the promoter regions of the S. cerevisiae SED1 or TDH3 genes (SGD:S000002484 and SGD:S000003424) and the terminator sequence of the DIT1 gene (SGD:S000002811) cloned onto pUC57 and separated by the recognition sequences for the restriction enzymes PacI and SgsI. To clone the relevant xylosidase and xylanase genes under the specified promoter (SED1P or TDH3P) and terminator (DIT1T), vector plasmids (pSED1p-DIT1t, pTDH3p-DIT1t) and plasmids containing the genes of interest (pRDH177_SED1, pRDH182) were individually digested with restriction enzymes PacI and SgsI (Thermo Fisher Scientific). The digested DNA was resolved on a 1% (w/v) agarose gel. After confirmation of size, DNA was extracted and purified from the agarose gel as done by Thuring et al. (1975) with modifications, and further purified using a standard phenol, chloroform, isoamyl alcohol (PCI) clean up. In brief, DNA bands were cut from the agarose gel and forced through a syringe, suspended in an equal volume of phenol, and frozen at − 80 °C for 45 min. After centrifugation at 13,000 rpm for 15 min, the upper phase was collected and combined with an equal volume of PCI (made up in a 25:24:1 ratio) and then vortexed. After further centrifugation at 13,000 rpm for 2 min, the upper phase was collected and combined with an equal volume of chloroform:isoamyl alcohol (CI) (made up in a 24:1 ratio) and vortexed. Samples were subsequently centrifuged at 13,000 rpm for 1 min and the upper phase collected and precipitated with 1/10 volume 3 M sodium acetate and 2 volumes of 100% ethanol at − 20 °C overnight. DNA was then pelleted and washed with 70% ethanol. After drying, DNA was resuspended in deionized distilled water.
The isolated vector and insert DNA was ligated with T4 DNA ligase (Thermo Fisher Scientific) according to the manufacturer’s instructions, after which ligated DNA was dialyzed against deionized distilled water on a 0.025-µm MCE membrane (Merck Millipore, Burlington, MA, USA). Dialyzed DNA was combined with electro-competent E. coli DH5α cells in a chilled electroporation cuvette (Bio-Rad, Hercules, CA, USA) and transformed via electroporation using a Bio-Rad MicroPulser (25 µF capacitance, 2.5 kV, and 200 Ohm resistance). Cells were quickly resuspended in super optimal broth with catabolite repression (SOC) media (20 g/L tryptone, 5 g/L yeast extract, 10 mM NaCl, 2.5 mM KCl) and incubated at 37 °C with shaking for 1 h. Cells were plated on LB agar supplemented with 100 µg/mL ampicillin and incubated at 37 °C overnight. To confirm successful ligation and transformation, resulting colonies were propagated in LB broth supplemented with 100 µg/mL ampicillin at 37 °C overnight on a rotary shaker and the plasmid DNA extracted using a Zyppy Plasmid Miniprep Kit as per the manufacturer’s instructions. Plasmid DNA was digested with PacI and SgsI restriction enzymes and resolved on a 1% agarose gel to determine if both the plasmid vector and gene insert were present. This way, the xln43_SED1 and xyn2 genes were both cloned into the pSED1p-DIT1t and pTDH3p-DIT1t plasmids to create four new plasmids (pSED1p-xln43_SED1-DIT1t; pTDH3pxln43_SED1-DIT1t; pSED1p-xyn2-DIT1t; pTDH3p-xyn2-DIT1t) (Table 1).
Strain construction
Constructed xln43_SED1 gene cassettes were PCR-amplified using forward primers Ch10_SED1p-L or Ch10_TDH3p-L, and reverse primer Ch10_DIT1t-R (Table 2), generating homology repair templates specific to an intergenic sequence in chromosome 10 (Mikkelsen et al. 2012) to be used in the subsequent CRISPR/Cas9-based yeast transformation. All PCR reactions made use of Taq DNA Polymerase Master Mix RED (Ampliqon, Odense, Denmark) according to the manufacturer’s instructions. PCR reactions were set up with 31 cycles of 95 °C denaturation for 30 s, 60 °C annealing for 30 s, and 72 °C elongation for 2 min and 30 s. Similarly, constructed xyn2 gene cassettes were amplified with forward primers Ch11_SED1p-L or Ch11_TDH3p-L, and reverse primer Ch11_DIT1t-R (Table 2) targeting chromosome 11 integration, with the only difference in the PCR reaction being the elongation time which was shortened to 2 min. PCR products were resolved on a 1% (w/v) agarose gel and purified using the PCI clean up. Purified DNA, as well as the plasmids containing CRISPR gRNA elements (pRS42-G-ChX, pRSCG_11; Table 1), was quantified on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and dialyzed against deionized distilled water on a 0.025-µm MCE membrane.
Table 2 Primers used in this studyThe S. cerevisiae S288C-MJM121 strain (M) used (Table 3) was a xylose-utilizing strain described by Mert et al. (2017). This strain was streaked out from a 15% (v/v) glycerol stock onto YPD agar (10 g/L yeast extract, 20 g/L peptone, 20 g/L glucose, and 20 g/L agar) and grown at 30 °C for 48 h. A single colony was inoculated into 20 mL YPD broth and incubated in an aerated conical flask at 30 °C at 180 rpm on an orbital shaker overnight. Yeast cells were harvested from the overnight culture and washed with deionized distilled water. To make the yeast cells electro-competent, washed cells were suspended in LiOAc/TE solution (100 mM LiOAc, 10 mM Tris–HCl pH 8.0, and 1 mM EDTA) and incubated at 30 °C at 180 rpm for 45 min. Subsequently, 20 µL 1 M dithiothreitol (DTT) was added to the mixture, which was incubated at 30 °C at 180 rpm for a further 15 min. The mixture was centrifuged, and the cells washed with distilled deionized water, then with electroporation buffer (1 M sorbitol and 20 mM HEPES). The yeast cells were transformed using approximately 10 µg of the respective DNA repair template and 1 µg CRISPR-based integration plasmid DNA with an electric pulse of 25 µF capacitance, 1.4 kV, and 200 Ohms. In this CRISPR/Cas9-based transformation, strains had the xln43_SED1 gene cassettes targeted to chromosome 10, and the xyn2 gene cassettes targeted to chromosome 11. Electroporated cells were resuspended in YPD broth supplemented with 1 M sorbitol. After both a 3-h and overnight incubation, the transformation mix was plated on YPD agar supplemented with 200 µg/mL Geneticin (G418) and incubated at 30 °C for 48 h.
Table 3 Yeast strains used in this studyConfirmation of successful transformation
Putative positive xln43_SED1 transformants were inoculated in 1.5 mL YPD broth in a 96-deep-well plate and sealed with a breathable AeraSeal™ film and incubated at 30 °C at 180 rpm for 48–72 h. These small-scale cultures were then screened for xylosidase activity on the substrate p-nitrophenyl-β-D-xylopyranoside (pNP-X). Cell culture was combined with 250 mM pNP-X and 50 mM NaOAc buffer in a 5:1:44 ratio to a 100 µL volume. This mixture was incubated at 50 °C for 30 min and the reaction was stopped by the addition of 100 µL 1 M sodium carbonate. Confirmation of positive xln43_SED1 transformants was indicated by a yellow colour formation.
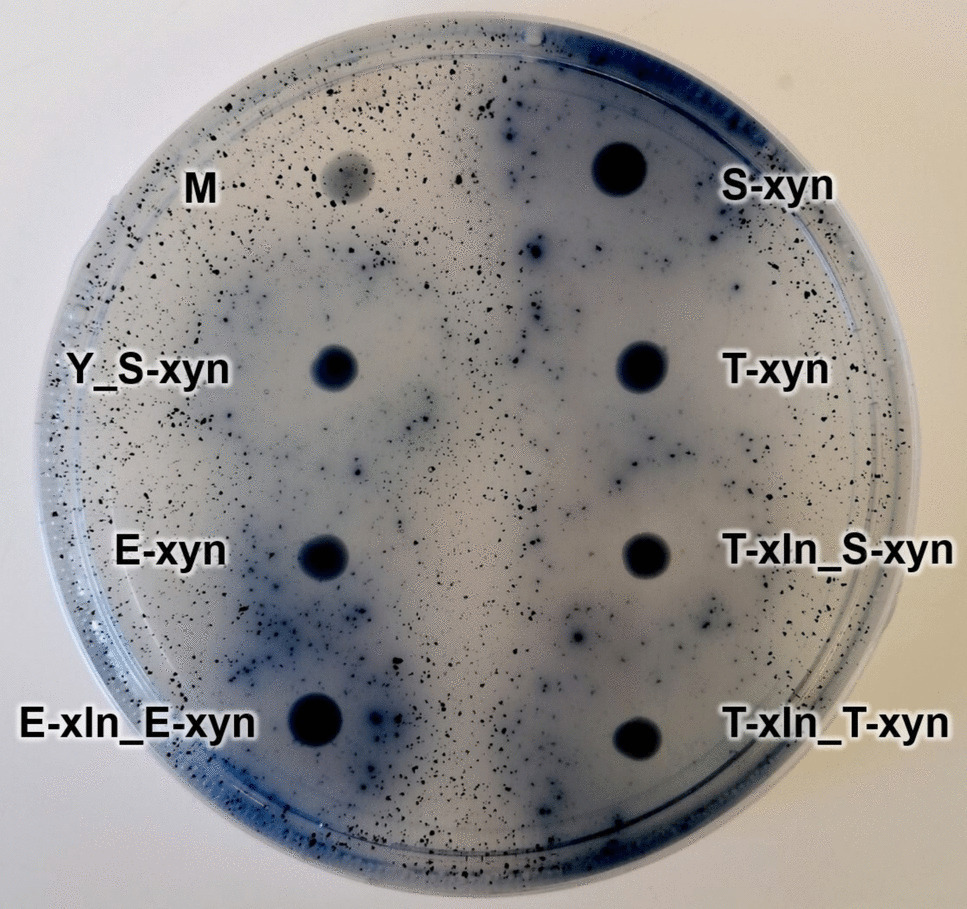
The confirmation of xyn2 transformants was done by spot plating putative transformant colonies onto SC−HIS agar (1.7 g/L yeast nitrogen base, 20 g/L glucose, 5 g/L ammonium sulfate, 1.5 g/L amino acids without histidine, and 20 g/L agar) supplemented with 1 g/L azurine cross-linked (AZCL)-xylan (Megazyme, Bray, Ireland) and incubating at 30 °C overnight. Positive xyn2 transformants were indicated by the presence of a dark blue halo surrounding the colony (Cedras et al. 2020). A previously constructed S. cerevisiae YI13 strain containing T. reesei xyn2 was used as a positive control. Notable strains constructed in this study were submitted to the publicly accessible Biobanks South Africa Yeast Culture Collection at the Department of Microbiology and Biochemistry, University of the Free State. Strain collection numbers are detailed in the supplementary material (Supplementary Table S1).
Enzyme activity assays
Overnight YPD pre-cultures of the engineered xln43_SED1 and xyn2 expressing strains were used to inoculate, in triplicate, 10 mL YPD in conical flasks or 20 mL YPD in rubber-stoppered bottles containing a glass bead to aid in culture mixing. Rubber-stoppered bottles were also pierced with sterilized 0.8 × 25 mm hypodermic needles stopped with cotton wool to allow for CO2 release while maintaining a micro-aerobic environment. Similarly, overnight YPX (10 g/L yeast extract, 20 g/L peptone, 20 g/L xylose) cultures of the engineered strains were made, which underwent an additional two sub-cultures before inoculating, in triplicate, into conical flasks with 10 mL YPX media. The sub-culturing was deemed necessary to allow sufficient growth and adaptation to xylose, which is not the preferred carbon source of the yeast strain used in this study. The YPD flasks, YPD rubber-stoppered bottles, and YPX flasks were incubated at 30 °C at 180 rpm for 72 h to determine xylosidase and xylanase enzymatic activity on glucose, under fermentative conditions, and on xylose, respectively.
The enzyme activity assays were all measured in triplicate. Measurement of cell-tethered xylosidase activity of the total cell culture was done using pNP-X substrate as outlined above in the preliminary screening with additional steps. After the addition of 1 M sodium carbonate, the mixture was centrifuged at 3000 rpm for 2 min, and 100 µL of the supernatant was transferred to a 96-well microtitre plate, and the absorbance was measured at 400 nm on a FLUOstar Omega Microplate Reader (BMG LABTECH, Ortenberg, Germany). The results were compared to a generated p-nitrophenyl (pNP) standard curve set between 0.075 mM and 1.25 mM to determine the amount of pNP released as a measure of xylosidase activity.
Evaluation of secreted xylanase activity was done using the dinitrosalicyclic acid (DNS) method as described by Bailey et al. (1992). Cell cultures were centrifuged at 3000 rpm for 2 min, and the supernatant was collected. The supernatant was combined with 10 g/L beechwood xylan (Megazyme) substrate (which had been made up to its final volume with 50 mM NaOAc buffer, boiled, and left to stir overnight) and incubated at 50 °C for 30 min. After the subsequent addition of DNS, the mixture was further incubated at 90 °C for 5 min, then 4 °C for 1 min. The final volume was 140 µL with a supernatant: substrate: DNS ratio of 1: 5: 8. Background sugar was determined by adding DNS to the supernatant before the addition of the xylan substrate to stop interaction between the xylanase enzyme and the beechwood xylan substrate, followed by the incubation step at 90 °C for 5 min, then 4 °C for 1 min. A 100 µL volume of each mixture was transferred to a 96-well microtitre plate, and the absorbance was measured at 540 nm spectrophotometrically on a FLUOstar Omega Microplate Reader. A standard curve was generated using xylose concentrations ranging from 0.5 g/L to 15 g/L. With high xylanase activity being observed in the cultures grown on YPD, the 50 °C incubation time was decreased to 5 min and supernatant diluted as necessary to stay in the range of the standard curve.
The OD600 of the grown cultures was measured to determine the DCW (g/L) of each culture (Jacob et al. 2022) to calculate enzyme activity as U/gDCW. The units of enzyme activity were expressed as U/L where one unit was defined as the amount of enzyme required to produce 1 µmol reducing sugar or equivalent per min (µmol/min). The calculated enzyme activity was then divided by the DCW (g/L) to get units of U/gDCW [(µmol/min)/L per gDCW].
Co-producing xylosidase and xylanase strain construction and enzymatic assays
Based on the results of the enzymatic assays, S288C-MJM121-based strains were constructed to contain both P. tritici-repentis xln43_SED1 targeted to chromosome 10, and T. reesei xyn2 to chromosome 11. These strains made use of what was determined to be the strongest tested promoter: TDH3P for both genes. Additionally, a second strain was constructed which made use of TDH3P (for the xln43_SED1) and SED1P (for the xyn2) to avoid potential overuse of TDH3P-related transcription machinery which may slow down the transcription process. These strains were constructed using the exact same methodology as stated above, except the host strain had been changed to a S. cerevisiae S288C-MJM121 strain (M-Cas) (Table 3) which was based on the same xylose-utilizing strain (Mert et al. 2017) and had been previously transformed with the pCas9-Nat plasmid to allow for CRISPR/Cas9-based engineering (Kruger and Den Haan 2022). M-Cas was streaked from a 15% (v/v) glycerol stock onto YPD agar supplemented with 100 µg/mL Nourseothricin (CloNAT) (Jena Bioscience, Jena, Germany). A single colony was then inoculated into 20 mL YPD broth supplemented with 100 μg/mL CloNAT and incubated in an aerated conical flask at 30 °C at 180 rpm on an orbital shaker overnight before electrotransformation (using the same methodology described above) with the constructed pTDH3p-xln43_SED1-DIT1t targeted to chromosome 10. The transformation mix was then plated on YPD agar supplemented with 100 μg/mL CloNAT and 200 μg/mL G418. After successfully screening for positive transformants using the previously described pNP-X assay, the newly constructed strain containing xln43_SED1 under TDH3P/DIT1T underwent 3–5 rounds of sub-culturing on YPD agar supplemented with 100 µg/mL CloNAT with 48 h in between each sub-culture to remove the remaining CRISPR-based integration plasmid DNA. The same CRISPR/Cas9-based methodology was used to insert the relevant xyn2 construct (pSED1p-xyn2-DIT1t or pTDH3p-xyn2-DIT1t; Table 1) into the yeast genome. Confirmation of successful transformation was done as previously stated using SC-based agar plates containing AZCL-xylan. This resulted in an additional two strains: T-xln_S-xyn and T-xln_T-xyn (Table 3). Strains containing both xln43_SED1 and xyn2 were then subjected to xylosidase and xylanase assays after cultivation on YPD, YPX, and under fermentative conditions. A strain containing the same genes, both under ENO1P/T, described in a previous report (Kruger and Den Haan 2022), was used as a benchmark strain against the newly constructed strains.
Growth trials
For growth trials on glucose- and xylose-containing media, strains were pre-cultured in their respective media (YPD or YPX), containing 100 µg/mL ampicillin and 100 µg/mL streptomycin (Thermo Fisher Scientific) to suppress bacterial growth, for 24 h at 30 °C and 180 rpm. The pre-cultures were used to inoculate flasks containing 10 mL YPD or YPX supplemented with 100 µg/mL ampicillin and 100 µg/mL streptomycin to an OD600 of 0.05. OD600 measurements were taken at regular intervals on a FLUOstar Omega Microplate Reader, and the resulting values converted to DCW (g/L).
To evaluate the growth on XOS and xylan substrates, strains producing both xylosidase and xylanase enzymes were pre-cultured in YPX media supplemented with 100 µg/mL ampicillin and 100 µg/mL streptomycin for 24 h. Following this, the strains were sub-cultured twice in fresh YPX media supplemented with 100 µg/mL ampicillin and 100 µg/mL streptomycin (Thermo Fisher Scientific) to an OD600 of 0.05 with 24 h in between each sub-culture.
Measurement of growth on XOS and xylan was done using the same methodology as described by Kruger and Den Haan (2022). To measure growth on XOS, pre-cultured strains were inoculated into conical flasks containing 10 mL SC−URA media without glucose supplemented with 50 g/L XOS (Carl Roth, Karlsruhe, Germany) to an OD600 of 0.05 and incubated at 30 °C shaking at 180 rpm. OD600 measurements were taken at regular intervals (every 24 h) over 16 days (384 h) on a FLUOstar Omega Microplate Reader, and the resulting values converted to DCW (g/L).
For growth on xylan, the same pre-cultured strains were inoculated into flasks containing 10 mL SC−URA media without glucose supplemented with 50 g/L beechwood xylan (Megazyme) to an OD600 of 0.05 and incubated at 30 °C shaking at 180 rpm. Samples were taken at regular intervals (every 24 h), diluted as necessary and counted under a compound light microscope using a Neubauer-improved haemocytometer counting chamber (Paul Marienfeld GmbH & Co. KG, Lauda-Königshofen, Germany), as described by Thomson et al. (2015), over 24 days (576 h). Cell counts were then converted to cells/mL before being converted to DCW (g/L).
The remaining culture of both the XOS and xylan growth trials was subjected to a phenol–sulfuric acid assay to determine the remaining XOS and xylan in the flask culture. Samples were centrifuged at 13,000 rpm for 10 min, and 200 µL of cell-free supernatant was combined with 200 µL 5% (w/v) phenol solution in a glass test tube in triplicate. Samples were vortexed gently, followed by the addition of 800 µL concentrated sulfuric acid and additional vortexing. 200 µL sample was transferred to a 96-well microtitre plate, and the absorbance was measured at 480 nm on a FLUOstar Omega Microplate Reader. XOS and xylan standard curves were generated using concentrations ranging from 0.5 to 8 g/L. The remaining sugar was calculated in g/L.
Xylan fermentation
The best-performing strain (T-xln_T-xyn), based on enzymatic assay results, was evaluated for the production of ethanol from beechwood xylan (Megazyme) in a CBP along with E-xln_E-xyn which served as a benchmark strain. YPX pre-cultures of these co-producing strains were grown for 24 h and were used to inoculate 50 mL YPX media supplemented with 100 µg/mL ampicillin and 100 µg/mL streptomycin to OD600 0.025. These cultures were incubated at 30 °C with shaking at 180 rpm for 96 h. Rubber-stoppered bottles containing 10 mL double-strength YP media (20 g/L yeast extract, 40 g/L peptone) supplemented with 80 g/L beechwood xylan (Megazyme) were inoculated in biological triplicate with 10 mL of the YPX flask culture (to achieve a final concentration of 40 g/L beechwood xylan). A sterilized glass bead was added to each bottle to improve the mixing of the fermentation broth. The rubber-stoppered bottles were pierced with sterilized 0.8 × 25 mm hypodermic needles plugged with cotton wool to allow for CO2 release while maintaining a micro-aerobic environment. The fermentation bottles were incubated at 30 °C with shaking at 180 rpm for 144 h, with 2 mL samples being taken at 0, 72, 120, and 144 h. These samples were centrifuged at 13,000 rpm for 10 min, and the cell-free supernatant was stored at − 20 °C until further analysis.
1 mL of the thawed supernatant was transferred to a clean Eppendorf tube and acidified with 50 µL 10% (v/v) sulfuric acid and vortexed briefly to ensure mixing. Samples were filtered through a 0.22-µm filter into 2 mL high-performance liquid chromatography (HPLC) vials. Ethanol, xylose, acetic acid, and glycerol concentrations were determined in each sample by an HPLC equipped with a Bio-Rad guard (part # 125–0129) 7.8 × 300 mm column and refractive index (RI) detector at a temperature of 65 °C with 5 mM sulfuric acid as the mobile phase at a flow rate of 0.7 mL/min.
Statistical analysis
All values obtained were presented as the mean of biological triplicates with their standard deviations. Significant differences between enzyme activities, growth data, and/or metabolite concentrations attained were investigated using two-tailed unpaired T-tests assuming unequal variance. A p-value < 0.05 was deemed significant. Calculations were done using Microsoft Excel.

Comments (0)