Yeast strains
B. mokoenaii CBS8435 (Y-27120) isolated from savanna soil in South Africa, B. illinoisensis CBS 10339 (YB-1343) isolated from a tree in Illinois, USA, and B. malaysiensis CBS 10336 (Y-6417) isolated from cave soil in Malaysia, were ordered from the ARS Culture Collection, Peoria, IL, USA (NRRL; https://nrrl.ncaur.usda.gov/). Strains were revived according to instructions from the provider and stored in 30% glycerol at − 80 °C.
Genome sequencing
B. illinoisensis and B. malaysiensis were grown in YPD overnight at 30 °C, 200 rpm, and cells were washed in milliQ purified water and harvested using centrifugation (6000 × g, 10 min). Cells were lysed using zymolyase-20T treatment at 37 °C, 30 min while buffered in 1 M sorbitol, 0.1 M ethylenediaminetetraacetic acid (EDTA)-Na2 at pH 7.5. DNA was extracted by adding 10 mL/g cell mix of 2% cetyltrimethylammonium bromide (CTAB) buffer (100 mM Tris–HCl, pH 8.0, 20 mM EDTA with 1.4 M NaCl), which was briefly vortexed and incubated at 57 °C for 1 h. DNA was extracted three times using phenol/chloroform and the 2-propanol precipitation method as described previously (Tõlgo et al. 2021). To remove residual RNA, genomic DNA was incubated with 200 µg mL−1 RNase A (Thermo Fisher Scientific, Waltham, MA, USA) at 60 °C for 2 h. Extracted genomic DNA was purified further using NucleoSpin soil kit (Macherey–Nagel, Düren, Germany), and DNA-RNA quality was analyzed using Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Approximately ~ 10 µg of high molecular weight genomic DNA was sent for PacBio NGS sequencing at SciLifeLab in Uppsala, Sweden. Library prep was performed using the SMRTbell Template Prep Kit according to the manufacturer’s instructions and sequenced on a single SMRT cell on a PacBio Sequel instrument (PacBio, Menlo Park, CA, USA).
Genome assembly and annotation
Whole-genome assembly was performed using the microbial assembly pipeline in SMRT Link v10 which uses HGAP4 for assembly and one round of polishing with arrow (Chin et al. 2013). Genome assembly statistics were computed using gfastats v1.3.6 (Formenti et al. 2022), and Benchmarking of Universal Single-Copy Orthologs (BUSCO) was performed using BUSCO v5.7.1 (Manni et al. 2021) with the saccharomycetes_odb10 gene set from OrthoDB v10 (Kriventseva et al. 2019). Prior to gene annotation, the polished assemblies were processed with RepeatModeler2 (Bourque et al. 2018) to generate a species-specific repeat library and then masked with RepeatMasker. Gene annotation was performed with Braker v3.0.3 (Gabriel et al. 2024), incorporating evidence in the form of all fungal proteins from OrthoDB v11 (Kuznetsov et al. 2023). The predicted genes were functionally annotated using the NBIS functional annotation nextflow pipeline v2.0.0 (https://github.com/NBISweden/pipelines-nextflow). Briefly, this pipeline performs similarity searches between the annotated proteins and the UniProtKB/Swiss-Prot database (Magrane and Consortium 2011) (downloaded on 2023–12) using BLAST. Then, it uses InterProScan (Jones et al. 2014) to query the proteins against InterPro (Paysan-Lafosse et al. 2023) v59-91 databases and merges results using AGAT v1.2.0 (https://doi.org/10.5281/zenodo.8178877). Transfer RNA (tRNA) and ribosomal RNA (rRNA) genes were annotated using tRNAscan-SE (Chan et al. 2021) v2.0.12 and barrnap v0.9 (https://github.com/tseemann/barrnap), respectively.
Comparative genomics
Pairwise average nucleotide identity (ANI) values were obtained from the whole-genome sequences of all species using fastANI (Jain et al. 2018) v1.34. OrthoFinder v2.5.5 was used to estimate the numbers of genes that could be clustered between species (Emms and Kelly 2019).
CAZyme prediction
Predicted protein-coding sequences from BRAKER v3.0.3 were used to predict CAZymes using the dbCAN3 server (https://bcb.unl.edu/dbCAN2/) (Zheng et al. 2023).
Yeast growth on different carbon sources
Yeasts were pre-cultured overnight at 30 °C, shaking 200 rpm in autoclaved liquid yeast extract–peptone–dextrose (YPD) containing 10 g L−1 yeast extract, 20 g L−1 peptone, and 20 g L−1 glucose. Growth on glucose, xylose, arabinose, galactose, lactose, and mannose (Sigma-Aldrich, Schnelldorf, Germany and Merck Rahway, NJ, USA); beechwood glucuronoxylan (Megazyme, Bray, Ireland); wheat arabinoxylan (Megazyme, Bray, Ireland); mixed-linkage β−1,3/1,4-glucan (barley, Megazyme, Bray, Ireland); glucomannan (konjac, Sigma-Aldrich, Schnelldorf, Germany); xyloglucan (tamarind, Megazyme, Bray, Ireland); and carboxymethyl cellulose (Sigma-Aldrich, Schnelldorf, Germany) was carried out in sterile Delft minimal media containing 5 g L−1 ammonium sulfate, 3 g L−1 potassium phosphate, 1 g L−1 magnesium sulfate, vitamins, and trace metals adjusted to pH 5 with 2 M KOH (Hendriks et al. 2018). After cell harvest and washing (4500 rpm, 5 min) of pre-cultures, yeasts were inoculated with a starting OD600 = 0.1 in liquid Delft media containing the different carbon sources at a concentration of 0.5–2% (w/v). Yeast growth in liquid cultures was monitored in triplicates over time at 30 °C and 200 rpm using a 96-well plate setup in a Growth-Profiler 960 (EnzyScreen, Heemstede, Netherlands). Agar plates containing 4 g L−1 (0.4%) carbon source and 20 g L−1 agar were inoculated with a 20 µL drop of cell suspension with a cell density of OD600 = 5 acquired from 3 × washed (8000 rpm, 10 min) 10 mL YPD yeast pre-cultures, spotted in the middle of the agar plate. Plates were kept at room temperature for 5 weeks while monitoring growth daily using an EPSON perfection V800 scanner (EPSON, Nagano, Japan) with a customized plate scaffold. The agar plate picture brightness was edited using the Affinity Photo 2 software (West Bridgford, UK).
Secretome activity in xylan-grown cultures
Yeasts were assayed for secreted xylanolytic activity over time from triplicates of 30-mL Delft media containing 20 g L−1 beechwood GX enriched liquid cultures using 100 µL samples which were centrifuged (10,000 × g, 5 min) at 4 °C and immediately frozen to − 20 °C after sampling. Endo-1,4-β-xylanase activity was quantified using a 250 µL mixture of thawed 25 µL cell-free supernatant mixed with fresh 10 g L−1 beechwood GX (Megazyme, Bray, Ireland) suspension buffered in 50 mM sodium acetate (pH 5). The buffered mixture was incubated for 30 min at 30 °C and 600 rpm followed by immediate chilling on ice and inactivation at 98 °C for 5 min. Reducing sugar contents were determined using the dinitrosalicylic acid method (McCleary and McGeough 2015). Secreted β-xylosidase activity from cell-free beechwood GX culture supernatants was quantified from 250 µL reactions containing 25 µL cell-free supernatant and 5 mM p-nitrophenyl-β-d-xylopyranoside buffered in 50 mM sodium phosphate (pH 7) in a 96-well plate and incubated for 30 min at 30 °C and 600 rpm. Spectroscopic quantification of p-nitrophenol was performed at 405 nm.
Secretomics and LC–MS/MS
Samples for secretome analysis were obtained from 30 mL Delft cultures with 20 g L−1 beechwood GX as sole carbon source, incubated at 30 °C, 200 rpm, and sampled after 72 h. Cell-free supernatants (12,000 × g, 5 min) were filtered through a 0.2 µm filter and frozen at − 20 °C until further processing. Samples were transferred to 15 mL 10 kDa Amicon spin columns (MilliPoreSigma, Burlington, MA, USA) and concentrated tenfold and buffer exchanged into sterile 50 mM sodium acetate buffer pH 5 with 50 mM NaCl. Protein concentration was measured using Nanodrop 2000 at 280 nm. Proteomics was performed by the Proteomics Core Facility, Sahlgrenska Academy, University of Gothenburg. Sample preparation was done using dl-dithiothreitol to reduce disulfide bonds and thereafter processed using the modified filter-aided sample preparation (FASP) method (Wisniewski et. al. 2009). Samples were analyzed by liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) using an Orbitrap Fusion Lumos Tribrid mass spectrometer with the FAIMS Pro ion mobility system, coupled with an Easy-nLC 1200 liquid chromatography system (Thermo Fisher Scientific, Waltham, MA, USA). Proteome Discoverer version 3.0 (Thermo Fisher Scientific, Waltham, MA, USA) was used for protein identification and relative quantification. The database search was performed using the Sequest search engine against custom databases with a precursor tolerance of 10 ppm and a fragment ion tolerance of 0.02 Da. Tryptic peptides were accepted with one missed cleavage; methionine oxidation was set as a variable modification, and cysteine carbamidomethylation was set as a fixed modification. Percolator was used for peptide spectrum matches (PSM) validation with a strict false discovery rate (FDR) threshold of 1%. Proteins were required to pass a protein FDR of 5%. LC–MS features were identified by the Minora Feature Detector node (Thermo Fisher Scientific, Waltham, MA, USA). Chromatographic alignment and feature mapping were enabled with a maximum RT shift of 5 min and a minimal signal-to-noise threshold of 5. Primary ion-intensity values at peak maximum for all unique peptides were used to calculate the corresponding protein abundances. Protein identification and analysis were processed using Pfam from the EGGNOG mapper available at www.galaxy.org, and signal peptides were predicted using the SignalP 6.0 online tool (https://services.healthtech.dtu.dk/services/SignalP-6.0/). Only proteins present in ≥ 2 of the three biological replicates, after filtering all search results to reach a protein FDR of 1%, were used for further analysis.
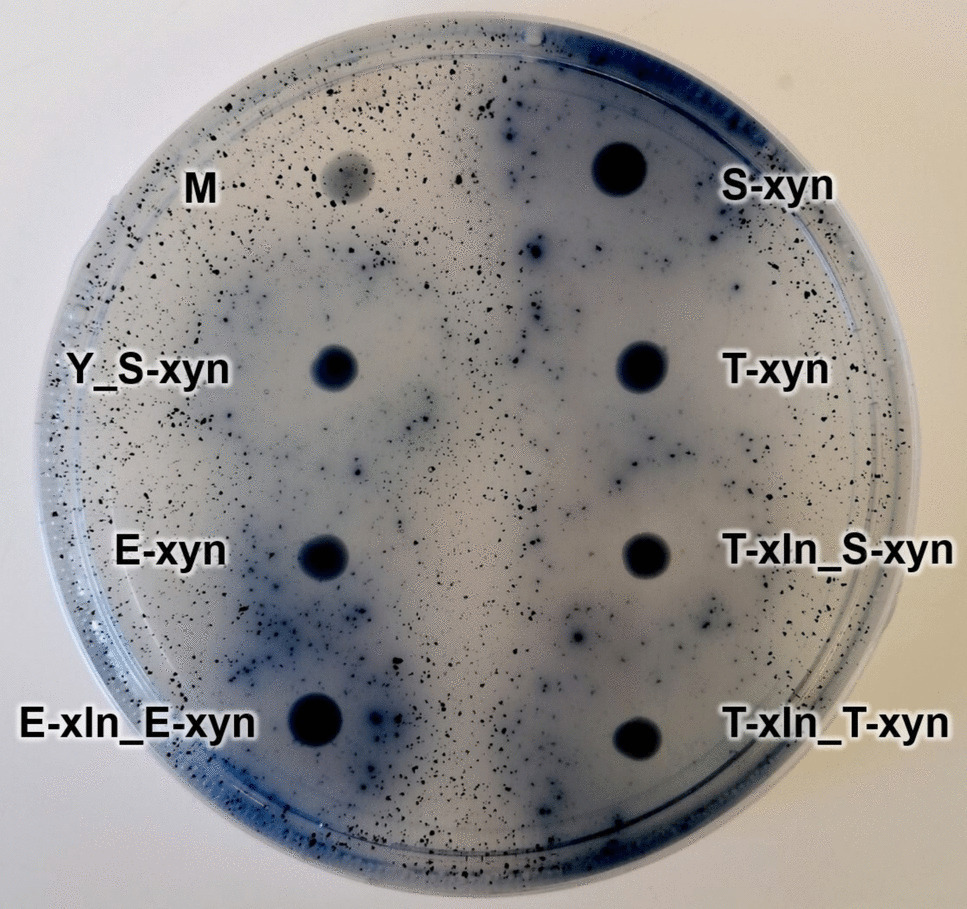
Zymogram analysis
Zymograms were produced by soaking pre-cast SDS-PAGE gels (Bio-rad, Hercules, CA, USA) loaded with 5–10 µL tenfold concentrated secretomes from the Blastobotrys beechwood GX enriched cultures in 100 mL 2% w/v Triton X-100 followed by 30-min shaking at 100 rpm twice to remove SDS. The protein gel was incubated in 100 mL of 0.1 M sodium acetate buffer at pH 5 for 15 min twice before soaking in 100 mL of 20 g L−1 beechwood GX buffered in 0.1 M sodium acetate at pH 5 for 60 min at 50 °C. After soaking with xylan, the gel was washed in 200 mL deionized water five times at room temperature at 60 rpm. The gel was subsequently stained with 100 mL 0.1% Congo red for 30 min and finally washed twice with 200 mL of 1 M NaCl for 15 min before fixing in 3% acetic acid and stored in the dark. Gels were visualized using regular photography on a plate with a backlight.
Heterologous protein expression
BmXyn30A (sequence ID in Supplementary List S1) was codon optimized and synthesized by GenScript (Rijswijk, Netherlands) after removal of the native signal peptide sequence for heterologous expression in Pichia pastoris X-33 and delivered in the pPICZa A vector (Invitrogen, Waltham, MA, USA). Recombinant pPICZa A constructs were coded for the yeast alpha-secretion factor, candidate gene, and a C-terminal His6 tag and were transformed into Escherichia coli DH5α One Shot Top10 cells (Invitrogen, Waltham, MA, USA). Transformants were selected using 25 mg mL−1 Zeocin in low-salt Luria–Bertani (LB) medium with 80 mM Tris–HCl at pH 7.5. The vector was propagated in 2 mL low-salt LB medium with 80 mM Tris–HCl, pH 7.5, with 25 mg mL−1 Zeocin and isolated using a GeneJET PCR purification kit (Thermo Fisher Scientific, Waltham, MA, USA). P. pastoris X-33 cells were transformed using 10 µg linearized (SacI restriction enzyme) and 96% ethanol precipitated pPICZa A using electroporation, and clones were selected using 100 µg mL−1 Zeocin on yeast extract–peptone–dextrose (YPD) plates containing 1 M sorbitol. Genomic integration of recombinant vectors were confirmed by colony PCR using primers aligning to the alpha-factor and AOX1 terminator plasmid parts. Positive P. pastoris X-33 clones were grown in medium scale at 20 mL, at 150 rpm and 25 °C in 100 mL baffled shake flasks in rich buffered glycerol-complex medium (BMGY). Methanol (1%, vol/vol) was used to induce expression in buffered methanol-complex medium (BMMY) over 4 days before harvesting as described in the EasySelect Pichia Expression kit (Thermo Fisher Scientific, Waltham, MA, USA).
Enzyme purification
Recombinant BmXyn30A and BmXyn11A proteins were purified by immobilized metal affinity chromatography (IMAC) using 2 × 5 mL Ni-Sepharose excel resin (GE Healthcare, Chicago, IL, USA) in gravity columns. After sample loading, the column was washed with five column volumes of loading buffer (50 mM Tris, pH 8, 250 mM NaCl) before elution of His6-tagged proteins with loading buffer containing 250 mM imidazole. Protein purity was evaluated by SDS-PAGE (Supplementary Fig. S1), and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) was used to determine protein concentration using the predicted molecular weights and extinction coefficients (Expasy ProtParam server, Swiss Institute of Bioinformatics).
Biochemical characterization of BmXyn30A and xylan degradation assays
BmXyn30A was biochemically characterized using a 200 µL mixture of 10 g L−1 beechwood GX (Megazyme, Bray, Ireland) in 50 mM sodium acetate buffer (pH 5) using 10 µL non-diluted purified enzyme with a specific enzyme concentration of 1.5 mg mL−1. The mixture was incubated for 60 min, with measurements taken at 0, 10, 20, 30, and 60 min, followed by immediate chilling on ice and inactivation at 98 °C for 5 min, before reducing sugars were quantified using the dinitrosalicylic acid (DNS) method, as described earlier (McCleary and McGeough 2015). For pH optimum measurements, either a 100 mM sodium citrate buffer with a pH range of 3–6 or a 100 mM sodium phosphate buffer with a pH range of 6–8 was used, and activity was quantified by the DNS method. Assays for the determination of the enzyme’s optimum temperature were carried out in 100 mM sodium acetate at pH 5, and reducing sugars were quantified by the DNS method.
Degradation assays were used to determine enzymatic additive effects in xylan hydrolysis, combining BmXyn30A and BmXyn11A with β-xylosidase GH43 (Xyl43) from Selenomonas ruminantium (cat. no. E-BXSR; GH43, Megazyme, Bray, Ireland) and α-methyl-glucuronidase BoAgu115A (Agu115) from Bacteroides ovatus (cat. no. CZO311; GH115, NZYTech, Lisbon, Portugal). A 1:1 molar ratio of BmXyn11A and BmXyn30A was used. Enzymes were incubated at normalized 0.1 µM concentrations in 400-µL mixtures of 10 g L−1 beechwood GX (Megazyme, Bray, Ireland) in 50 mM sodium acetate buffer, pH 5, for 16 h at 40 °C and 650 rpm, and then inactivated for 5 min at 98 °C before analysis by the DNS method.
Xylooligosaccharide analysis by ion chromatography
The formation of xylooligosaccharides from enzymatic hydrolysis of 10 g L−1 beechwood GX (Megazyme, Bray, Ireland) by BmXyn30A and BmXyn11A combined with Agu115 and Xyl43 was analyzed after 16 h reactions (0.1 µM enzyme concentration in 10 g L−1 beechwood GX and 50 mM sodium acetate pH 5, at 40 °C and 650 rpm in 400 µL total volume). Samples were inactivated at 98 °C for 5 min before supernatants were filtered through a 0.2 µm filter and diluted twofold in sterile water and stored at 4 °C. Analysis was performed using a high-performance anion-exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD) and an ICS-5000 system (Dionex Sigma-Aldrich, Sunnyvale, CA, USA), operating at 25 °C. A CarboPac PA200 (250 mm by 3 mm) column (Thermo Fisher Scientific, Waltham, MA, USA) was used for separation of hydrolysis products with a gradient of eluents using a flow rate of 0.5 mL min−1: A, milliQ water; B, 300 mM sodium hydroxide; and C, 100 mM sodium hydroxide and 1 M sodium acetate. Standards (5 to 800 µM) of xylose (X1), xylobiose (X2), xylotriose (X3), xylotetraose (X4), xylopentaose (X5), and xylohexaose (X6) (Megazyme, Bray, Ireland) were used for quantitation. Xylobiohydrolase activity by BmXyn30A was analyzed using 200 µM xylotetraose in 50 mM sodium acetate buffer, pH 5, with time-course measurements taken at 0, 30, 60, 90, and 120 min, which were inactivated by 5 min incubation at 98 °C prior to HPAEC-PAD analysis.

Comments (0)