
Remember me
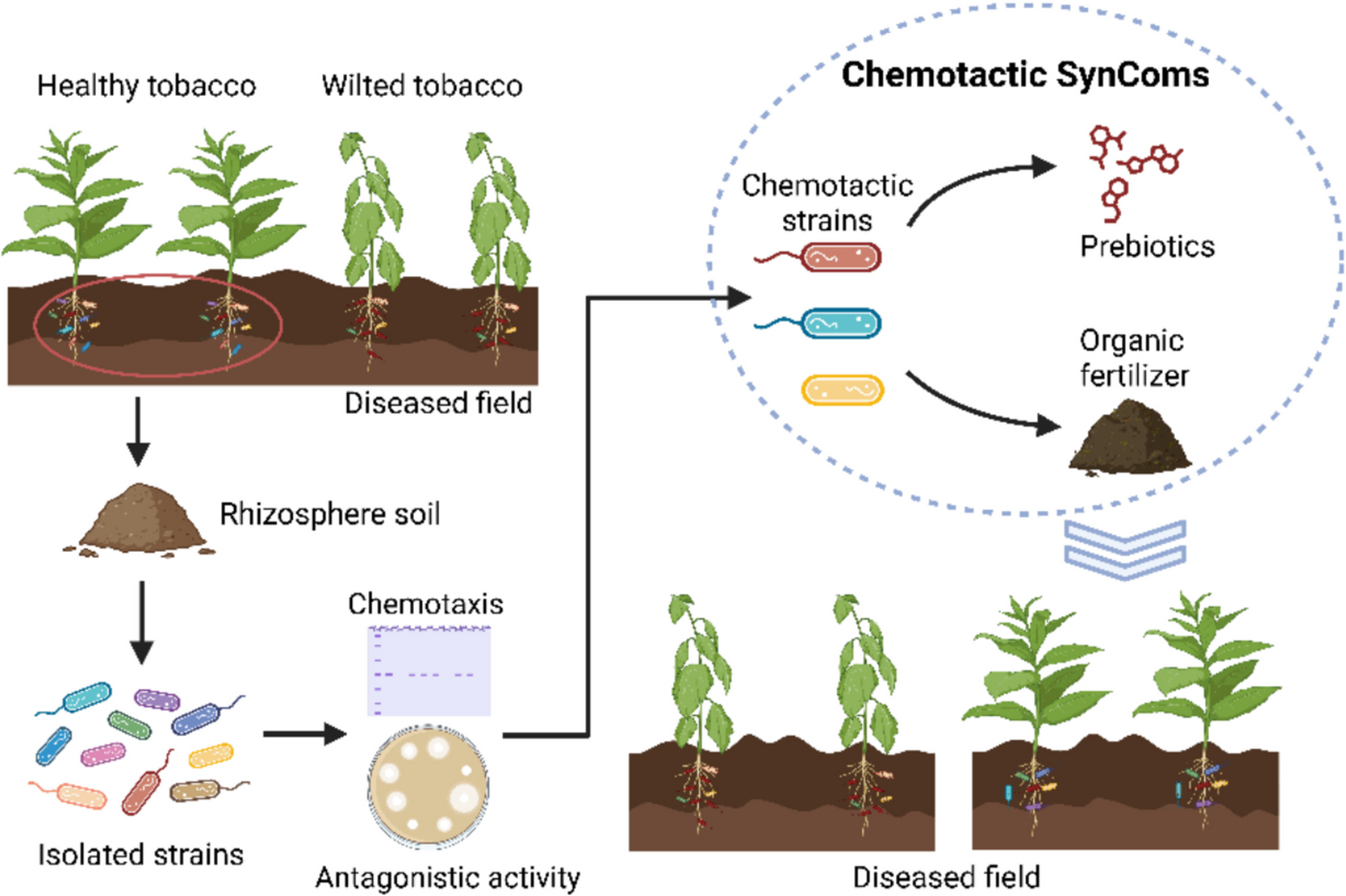
Preliminary characterization studies demonstrated that Sbv333-ATA has a pH optimum of 9.0, it is extremely thermostable, retaining 100% of starting activity after 3 h of incubation at 70 °C and has a melting temperature of 85 °C (Ferrandi et al. 2021). The Sbv333-ATA stability was further tested in the presence of 5–20% (v/v) of water-miscible cosolvents: methanol, ethanol, dimethyl sulfoxide and acetonitrile. To assess the stability, the activity was evaluated using the acetophenone assay, as described in the “Materials and methods” section, at the beginning of the experiment and after 5 and 24 h. The biocatalyst proved to be stable in up to 20% (v/v) of organic solvent, retaining at least 40% of the starting activity after 24 h (Fig. 1a, Table S3).
Fig. 1
Sbv333-ATA stability to organic solvents. a Stability to water-miscible cosolvents, residual activity after 5 h and 24 h of incubation with 5–20% (v/v) of different solvents (methanol, MeOH; ethanol, EtOH; dimethyl sulfoxide, DMSO; acetonitrile, ACN). b Stability in biphasic systems (1:1), residual activity after 5 h and 24 h of incubation under shaking with ethyl acetate (EtOAc), petroleum ether (PE), and toluene (Tol) as organic phase. Residual activities were calculated considering as 100% the enzymatic activity before solvent addition (see Supplementary Information, Tables S3 and S4 for details). Reactions were performed at least in triplicate; standard deviation of residual activity was below 5%
The stability of Sbv333-ATA was also tested in biphasic systems (1:1, v/v), with either ethyl acetate, petroleum ether, or toluene as the organic phase. The biphasic mixtures were maintained under vigorous shaking, and the residual activity in the aqueous phase was evaluated using the acetophenone assay after 5 and 24 h. As shown in Fig. 1b (Table S4), Sbv333-ATA was tolerant to the different solvents, retaining more than 50% of its activity even after 24 h of incubation.
To evaluate the substrate scope of Sbv333-ATA, a series of aliphatic and aromatic amines, as well as different amino acids (Fig. 2a), were assayed as potential amine donors using the glycine oxidase (GO) assay (see the “Materials and methods” section for details). The enzyme was active toward a diverse set of substrates, including (S)-methylbenzylamine ((S)-1), 2-phenylethylamine (7), propylamine (10), cadaverine (11), and selected amino acids, while, as expected, more sterically hindered aromatic amines were not accepted (Fig. 2b). Negligible results were obtained with isopropyl amine, a commonly used amine donor. The substrate (S)-1-phenylpropylamine ((S)-2) precipitated in the assay solution, making it impossible to assess enzyme activity toward this substrate with this method.
Fig. 2
Amino donor spectrum of wild-type Sbv333-ATA. a Panel of screened amines; b Sbv333-ATA specific activity (mU mg−1) toward accepted amines and amino acids. Reactions were performed at least in triplicate; standard deviation of residual activity was below 5%
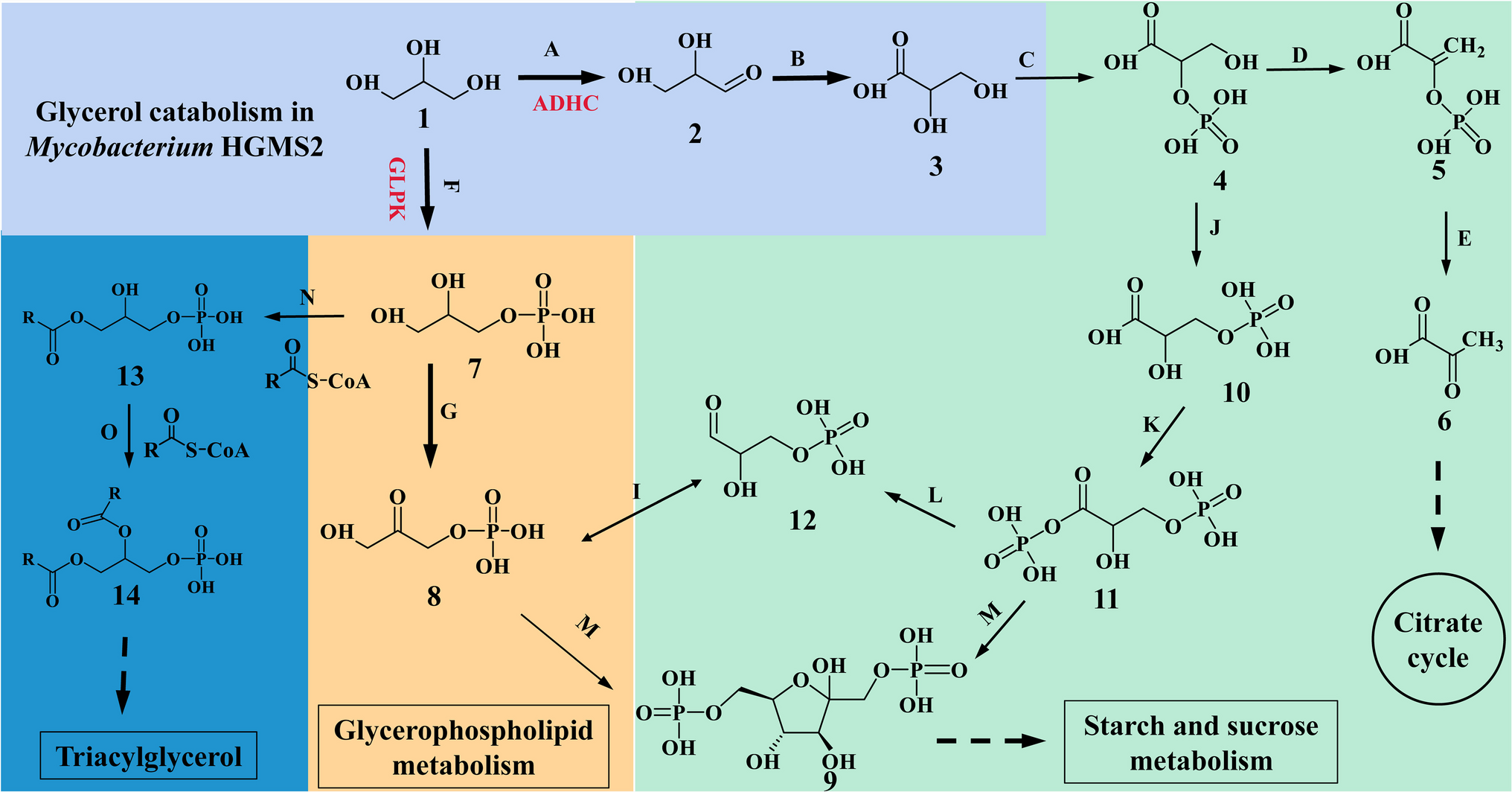
Sbv333-ATA 3D-structure determination and analysisThe structure of wild-type Sbv333-ATA was determined and refined to a resolution of 1.49 Å.
This enzyme crystallized as a homotetramer in the asymmetric unit. Analytical gel filtration chromatography yielded a single peak corresponding to an apparent molecular weight of 100 kDa, which would indicate a dimer. Crystals were grown without the addition of any PLP to the crystal droplet, and they grew to dimensions of 0.2 mm. A crystal of this form diffracted to 1.18 Å and belonged to the space group C 2 2 21, with the following unit cell parameters: a: 100.00, b: 176.96, c: 111.93, α = β = γ: 90.00.
The Sbv333-ATA monomer structure exhibits the typical transaminase type I fold (Van Oosterwijk et al. 2016), consisting of two main domains (Fig. 3). The large domain (residues 72–345) binds the PLP cofactor and contains a mostly parallel seven stranded β-sheet flanked by α-helices on both sides. The small domain comprises residues 1–71 and 346–459. The latter part contains a three-stranded antiparallel β-sheet flanked by α-helices. The N-terminal tail (residues 1–37) forms a long loop and an α-helix and is involved in the dimer contacts with the α-helix 3 and β-sheet 4 as well as a long loop consisting of residues 313–323.
Fig. 3
The Sbv333-ATA dimer with one monomer (right) shown with the large and small domain in dark blue and brown respectively and the PLP cofactor shown as a sphere model, the N-terminal tail and loop involved in the dimer contacts are shown in gold, and the second monomer (left) in light blue
The PLP cofactor is covalently attached to the Lys289 side chain via a Schiff base. It binds in a pocket lined with aromatic residues (Fig. 4), with all its non-carbon atoms forming hydrogen bonds with the surrounding residues from both monomers in the dimer.
Fig. 4
a The cofactor binding pocket of Sbv333-ATA, the Lys289–PLP Schiff base is highlighted as a green stick model, surrounding residues are shown in gold and light blue to discern the two opposing monomers. The dotted lines represent stabilizing hydrogen bonds. b Quality of the electron density at the Lys289–PLP Schiff base contoured at 1.0 σ
Similar to most transaminases, the Sbv333-ATA active site consists of two pockets for substrate interaction, the large (L) and the small (S) pocket. The active site cleft is formed by the two domains of one monomer and the large domain of the neighboring monomer. While residues from both subunits are involved in cofactor binding, the substrate site is primarily formed by residues from the two domains of one monomer.
Gabaculine-inhibited complexGabaculine is widely recognized as a suicide inhibitor of aminotransferases. It binds to the enzyme and forms a Schiff base with the PLP cofactor. The inhibition process involves initial proton abstraction followed by a second proton removal from the adjacent carbon atom, creating an unstable intermediate. This intermediate is then converted to m-carboxyphenylpyridoxamine phosphate (mCPP), a highly stable compound that results in an irreversible aromatic modification of the cofactor (Rando 1977; Sayer et al. 2012). This inhibitor allows the enzyme to “freeze” in a state that resembles that of the substrate bound state. The electron density in the enzyme active site allowed modeling of the cofactor covalently bound to gabaculine as the irreversible mCPP ligand. A 2Fo-Fc electron density map for the mCPP ligand is shown in Fig. 5a.
Fig. 5
a Electron density map of the gabaculine complex calculated at 1.5 Å resolution. The nonhydrolysable PLP–gabaculine complex is shown as an mCPP molecule. b Surface view of the large pocket (green) featuring residues F23, L166, L169, and D421 depicted as stick models, and the small pocket (blue) containing residues L60, F61, Y153, N231, and W89. The mCPP molecules (orange) occupy the small pocket, leaving a visibly large empty area in the large pocket above
The mCPP is positioned centrally in the active site in close proximity (< 4.5 Å) to the S pocket. The gabaculine part of the mCPP molecule is stabilized by H-bonds with N231, R419, and R423, and by hydrophobic interactions with L60 and F61, while the PLP part is bound by hydrogen bonds with G120, G121, and D260, and is held in place by hydrophobic interactions with residues Y153, H154, Q227, and V262 (Fig. S6, Supplementary Information).
On the opposite side of the active site, the L pocket outlined by residues F23, L166, L169, and D421 forms a much larger cavity which could bind the large aromatic group of substrates (Fig. 5b) (Sayer et al. 2014).
Native Sbv333-ATA has also been co-crystallized with the substrate phenylacetylcarbinol (PAC) and the product analogue norephedrine (see chemical structures in Fig. S1) (Fracchiolla et al. 2023; Patti et al. 2024). X-ray data were collected at 1.29 Å resolution for crystals of these two complexes. Electron density in both structures clearly shows two conformations of the main chain for residues 419–426, with observed displacement of Cα atom positions of up to 10 Å. One of the alternative main chain conformations exactly matches that observed in the native Sbv333-ATA structure. The other conformation differs from that observed in the gabaculine complex and has not been previously reported in any omega/amine TA structure.
The electron density of the bound ligands is partially masked by the alternative conformation of the main chain, complicating refinement of these complex structures. Despite the high concentration of co-crystallized compounds (10 mM), which should have resulted in full occupancy of the ligands in the active site, only partial occupancy was observed. This partial occupancy may result from allosteric properties of Sbv333-ATA.
Protein engineeringThe Sbv333-ATA, as demonstrated by studies shown in this paper and in our previous work (Ferrandi et al. 2021), is an enzyme with interesting features of thermostability and stability to organic solvents. Therefore, it was chosen as a starting point for the rational design of new variants with broader substrate scope, thus expanding the toolbox of stable transaminases able to interact with bulky substrates that can be used as pharmaceutical building blocks. Moreover, we also tried to reverse the enzyme’s enantioselectivity.
From the analysis of the gabaculine bound crystal structure described above, six amino acidic positions not essential for the catalytic mechanism and which contribute to the shape of the active site pockets were identified within a 5 Å radius from the bound gabaculine (F23, L60, F61, W89, Y153 and D421). Therefore, by replacing these positions with smaller amino acid residues, it is, in theory, possible to enlarge the small pocket and, in this way, allow bulky substrates to enter the active site and be converted.
Particularly, mutants L60A, L60V, W89A, W89Y, F61C, F61V, F61A, and W89A/L60V were designed with the aim of enlarging the small pocket, while mutant F23V was designed to promote bulky substrate binding by enlarging the large pocket. Mutant Y153W and the double mutants W89Y/Y153W, W89A/F23W, W89A/D421E, W89A/D421W, and W89A/F61W were designed to invert the reaction’s stereoselectivity. This could in principle be achieved by either simultaneously enlarging the small pocket and constricting the large pocket (in the case of double mutants) or by introducing a steric clash with the substrate’s aromatic ring (as with mutant Y153W) to alter enantioselectivity.
Expression trials of variants carrying up to two-point mutations were carried out, and 10 out of 14 mutants were successfully overexpressed in soluble form (see Table S2 for details).
The mutants designed to bind bulkier substrates, L60V, W89A, F61C, F61V, F23V, and W89A/L60V were characterized based on their amine donor spectrum. Screening was performed using the previously described glycine oxidase (GO) assay and the same panel of amines tested for the wild-type Sbv333-ATA (Fig. 2, Table S5). This analysis aimed to evaluate their activity on bulky substrates, such as (S)-1-phenylbutylamine ((S)-3) and 1,2-diphenylethylamine (4), while also assessing whether they retained activity on other substrates accepted by the wild-type enzyme.
Under the assay conditions used, variant F61V was completely inactive, while F23V, L60V, and W89A/L60V displayed only minimal activity toward the tested substrates. However, some promising mutants with an expanded substrate scope were identified (Fig. 6, Table S5). In particular, the F61C and W89A variants retained a substrate specificity comparable to that of the wild-type enzyme while also exhibiting the ability to accept new substrates. F61C could convert substrate (S)-1-phenylbutylamine ((S)-3), and W89A could accept both substrates (S)-3 and 1,2-diphenylethylamine (4).
Fig. 6
Amine donor screening of Sbv333-ATA variants. Assays were performed using the glycine oxidase (GO) assay (Weiß et al. 2014)
Interestingly, the structure of W89A was solved at 1.24 Å resolution and a substantial enlargement of the small pocket was observed, making the enzyme capable of accepting the bulky substrate 4, as shown in Fig. 7a. Moreover, computational molecular docking of the aromatic substrate 4 into the active site of the W89A mutant revealed a docking pose of 1,2-diphenylethylamine in a catalytically productive orientation, in agreement with the activity data (Fig. 7b).
Fig. 7
a Superposition of wild-type Sbv333-ATA (orange) and W89A variant (light blue) highlighting the enlargement of the S pocket resulting from the mutation of tryptophan 89 to alanine. The bulk of tryptophan 89 is shown as surface representation using the same color scheme. b Computational docking of 1,2-diphenylethylamine (4) into Sbv333-ATA W89A. The catalytically productive orientation of 1,2-diphenylethylamine is shown in orange sticks. c Superposition of wild-type Sbv333-ATA (orange) and F61C variant (light blue) highlighting the enlargement of the S pocket resulting from the mutation of phenylalanine 61 to cysteine, using the same color scheme as in panel a. d Computational docking of 1-phenylbutylamine ((S)-3) into Sbv333-ATA F61C. The catalytically productive orientation of 1-phenylbutylamine is shown in pink sticks
In a similar approach, the crystallographic structure of the F61C variant was determined at 1.31 Å resolution (Fig. 7c). The substitution of phenylalanine 61 with the smaller cysteine residue effectively reduced steric hindrance in the active site, as intended. This structural modification allowed for molecular docking of bulky phenylbutylamine ((S)-3) in multiple catalytically favorable conformations, with the lowest energy pose depicted in Fig. 7d.
In the study on stereoselectivity switching, the mutants Y153W, W89Y/Y153W, W89A/D421E, W89A/D421W, and W89A/F61W were tested against selected aromatic (R)-amines with increasing side chain lengths, namely, (R)-methylbenzylamine ((R)-1), (R)-1-phenylpropylamine ((R)-2), and (R)-1-phenylbutylamine ((R)-3) (see chemical structures in Fig. S2) using the GO assay. Unfortunately, all mutants were inactive toward these substrates.
To further assess their activity with (S)-amines and other amine donors, the mutants underwent a substrate screening using the GO assay with the compounds shown in Fig. 2. Mutants W89A/D421W and W89A/F61W were completely inactive with all substrates, whereas mutants W89Y/Y153W and Y153W exhibited low activity. Interestingly, the W89A/D421E variant retained full activity, displaying a substrate scope and specific activity largely comparable to that of W89A (Fig. S3).
Biotransformation with bulky substratesTo confirm the results obtained with the GO assay with (S)-methylbenzylamine ((S)-1) and the bulky substrate (S)-1-phenylbutylamine ((S)-3), and to verify the activity toward (S)-1-phenylpropylamine ((S)-2) (which, as previously mentioned, precipitated in the GO assay solution), reactions with the expressed mutants were carried out on an analytical scale using pyruvate as the amine acceptor. The reactions were monitored by GC/MS analyses. The wild-type enzyme was active in the deamination of (S)-2 and, as expected, demonstrated a decrease in enzyme activity when increasing the amine side chain.
Unlike the results obtained with the GO assay, mutants F23V and L60V showed slight activity toward substrates (S)-1 and (S)-3, respectively. In general, the other findings from the GO assay were corroborated, and among all the tested mutants, as expected, W89A stood out as the most promising, demonstrating the ability to catalyze the quantitative conversion of substrates (S)-1, (S)-2 and (S)-3 (Fig. 8).
Fig. 8
Biotransformation of selected aromatic bulky amines with increasing side chain catalyzed by wild-type Sbv333-ATA and selected mutants. Reactions were performed at least in triplicate; standard deviation of residual activity was below 5%
Among the other mutants, F61C displayed slightly lower activity than the wild-type enzyme toward substrate (S)-1, comparable activity with substrate (S)-2, and a fourfold higher conversion with substrate (S)-3. On the other hand, the L60V mutant exhibited lower activity across all three substrates when compared to the wild type. Additionally, the double mutant W89A/L60V was less active than W89A alone.
Mutant W89A, which demonstrated activity toward the bulky-bulky substrate 1,2-diphenylethylamine (4) in the colorimetric screening, was also tested in small-scale biotransformation with optically pure (S)-4. Remarkably, it exhibited high substrate conversion (> 90%), confirming its activity on this sterically hindered molecule.
Finally, mutant W89A was tested in small-scale biotransformation of (R)-amines ((R)-methylbenzylamine ((R)-1), (R)-1-phenylpropylamine ((R)-2), (R)-1-phenylbutylamine ((R)-3), and (R)-1,2-diphenylethylamine ((R)-4), see chemical structures in Fig. S4) to verify its stereospecificity. Considering that for the wild-type enzyme, increased pH values were associated with enhanced conversion rates, but at the expense of enantioselectivity (Patti et al. 2024), reactions were performed at pH 9.0 or at pH 7.5 for comparison. As shown in Fig. S4, the W89A variant showed low conversions at pH 9.0 (< 20%), while at pH 7.5 the enzyme exhibited higher stereoselectivity, with only some residual activity on (R)-1. Thus, the trend observed with the W89A mutant aligns with our previous findings on the wild-type enzyme, i.e., confirming its (S) stereospecificity and highlighting the effect of pH in the outcome of (R)-amines deamination reactions.
Comments (0)