Targeting lipid metabolism within the tumor microenvironment, particularly through LDs, has the potential to reduce chemoresistance and improve the effectiveness of standard therapies. In this study, we explored how non-anticancer drugs (DEX, CXB, and SMV) modulate 5-FU resistance via LD accumulation.
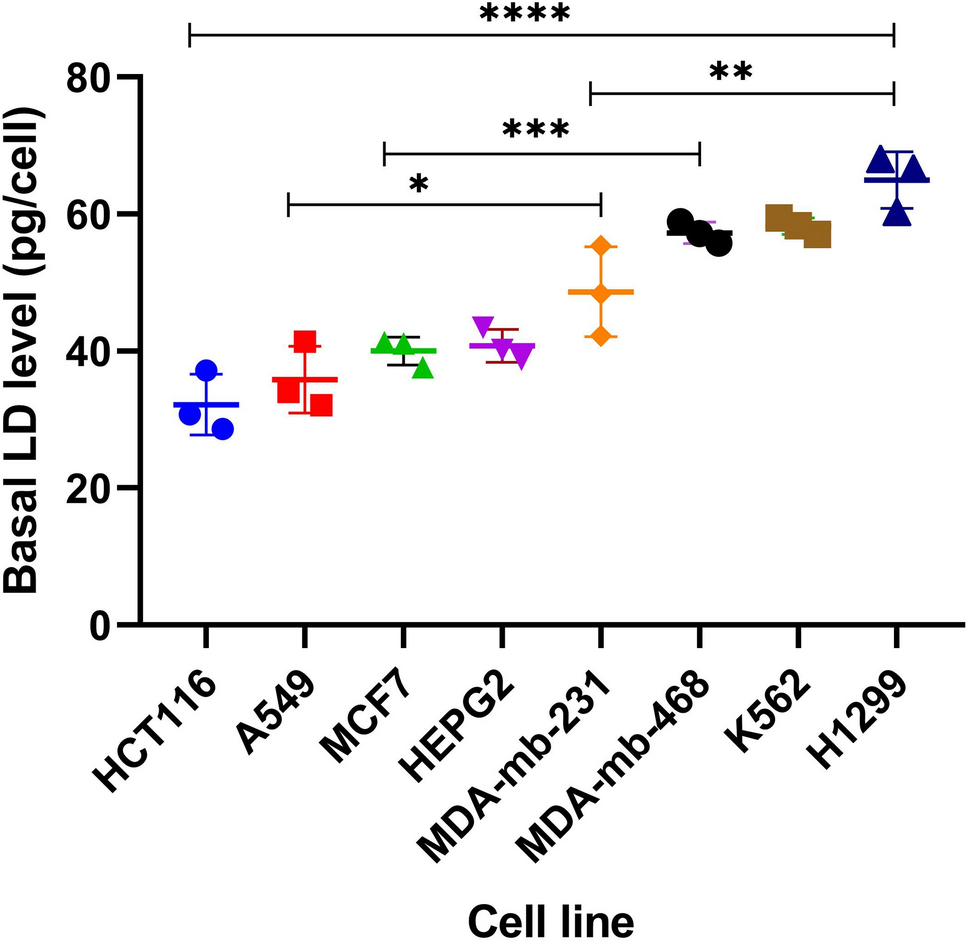
Building on previous research regarding LDs' role in chemoresistance, we examined whether basal LD levels can predict cancer cell responses to chemotherapy. Analyzing eight cancer cell lines revealed a moderate positive correlation between basal LD levels and 5-FU IC₅₀ values (r2 = 0.6185), which was confirmed with laboratory-derived IC₅₀ values (r2 = 0.5235) (Supplementary Table 1, Fig. 2). Previous findings by Cotte et al. support this, showing that elevated basal LD levels correlate with 5-FU resistance in CRC cells [7]. Furthermore, Germain et al. discussed how LDs contribute to therapy resistance by influencing lipid metabolism, signaling pathways, and cellular stress responses [12]. These findings underscore the potential of LDs as biomarkers of chemoresistance; however, their variability across tumor types and the dynamic nature of LD levels challenge their predictive value in clinical settings.
For several decades, 5-FU has been extensively used in treatment regimens against many types of malignancies [13]. 5-FU exerts its cytotoxic effects primarily by inhibiting thymidylate synthase (TS) or by incorporating its metabolites into RNA and DNA, thereby disrupting biosynthetic processes within cancer cells [13]. Despite its efficacy, the clinical utility of 5-FU is often compromised by resistance mechanisms—not only through TS overexpression but also via alterations in lipid metabolism, autophagy, apoptosis, and oxidative stress pathways [9]. Recent studies have highlighted that sequential drug treatments can induce epigenetic changes; for instance, in response to doxorubicin and paclitaxel in breast cancer cells [14]. LDs are emerging as sites for the regulation of different signaling pathways with potential functions in the tumor microenvironment and chemoresistance [5]. Tamoxifen-resistant breast cancer cells exhibit lipid reprogramming characterized by increased cholesterol production and LD accumulation [8]. In CRC, elevated LD content and lysophosphatidylcholine acyltransferase 2 (LPCAT2) expression correlate with resistance to 5-FU and oxaliplatin [7]. These findings suggest that targeting lipid metabolic reprogramming, either by using LDs as biomarkers or by diminishing LD biogenesis/function, could improve patient outcomes, whereas combining 5-FU with LD-elevating drugs may attenuate its response [15].
Our data also show that exposure to 5-FU increases LD levels in all tested cell lines (MDA-MB-468, A549, and HCT116) (Fig. 3, Supplementary Table 2). Similarly, Mehdizadeh et al. reported that treatment with doxorubicin and 5-FU induces the formation of highly saturated LDs and alters membrane lipid composition, potentially enhancing membrane fluidity and promoting invasion and metastasis [16]. Notably, the elevation of LD levels after 5-FU exposure varied among the three tested cell lines [Fig. 3]. A549, with a low basal LD level (35.83 ± 4.86 pg/cell), showed the highest increase in LD levels after 5-FU exposure (70.17 ± 1.43 pg/cell) (***p < 0.001). Conversely, MDA-MB-468, with a high basal LD level (57.26 ± 1.47 pg/cell), showed a lower elevation in LD accumulation after 5-FU treatment (70.14 ± 3.60 pg/cell) (*p < 0.05). MDA-MB-468 cells exhibit inherently high basal LD levels, which may limit their capacity for further LD accumulation upon treatment, potentially contributing to their distinct response. Additionally, it remains unclear whether treatment-induced changes in LD composition, rather than quantity, play a role in chemoresistance. Notably, in A549 cells, LD quantity did not correlate with resistance, suggesting that factors beyond LD accumulation, such as lipid composition or metabolic adaptations, may influence drug sensitivity. Further investigation is needed to elucidate these mechanisms. In contrast, HCT116, which is considered to have a low basal LD level (32.18 ± 4.43 pg/cell), showed a lower elevation in LD accumulation after 5-FU treatment (45.83 ± 1.88 pg/cell). These results are consistent with findings that indicate differential LD responses to chemotherapeutic stress among different cancer types [16]. However, these results contradict the findings of Cotte et al., where basal LD levels in CRC cells were negatively correlated with the proportion of LD that increased after 5-FU treatment [7]. The suggested negative correlation in the study performed by Cotte et al. was based on results obtained from two cancer cell lines from the same origin, CRC cells (SW620, HT29), which had different sensitivities to 5-FU (10 µM, 400 µM) respectively [7]. This may suggest poor representation of the LD accumulation process. Moreover, it is expected that resistant cells, such as HT29 (IC50: 400 µM), will not be under stress when exposed to a low 5-FU concentration (10 µM); therefore, LD levels, which are considered a stress marker, will not increase significantly. In our study, selected cell lines (HCT116, A549, MDA-MB-468) showed similar high chemosensitivity to 5-FU at low concentrations (Supplementary Table 2). Selecting cells with high chemosensitivity and similar IC50 values increases the probability of 5-FU inducing its activity with the same mechanisms of action on these cells. 5-FU may act differently when cells are exposed to high concentrations, as in such conditions off-target hits with less affinity to 5-FU may be involved, increasing the chance of additional mechanisms leading to cell death.
The 5-FU-induced LD increase, despite no clear correlation, suggests cell-specific factors like LD composition and adaptive mechanisms. Various studies have explored adjuvant and neo-adjuvant 5-FU regimens with anticancer and non-anticancer agents to enhance treatment efficacy. [17, 18]. Recently, it has been reported that antitumor response rates improved between 40 to 50% when 5-FU was administered in combination with oxaliplatin and leucovorin in patients with colorectal or gastrointestinal cancers [17]. Interestingly, oxaliplatin’s anticancer activity is reported to be mitigated by elevated LD levels in cancer cells [7], This raises whether 5-FU-induced LDs limit treatment benefits. Similarly, palliative therapies targeting lipid metabolism may counter chemoresistance and enhance response [19]. However, their role in overcoming resistance remains underexplored.
Non-anticancer drugs like CXB, DEX, and SMV, used in palliative care, have been reported to improve the overall well-being of cancer patients. CXB, is the optimum non-steroidal anti-inflammatory drug (NSAID) choice for cancer patients experiencing pain [20]. DEX, is also used at high doses to prevent chemotherapy-induced nausea and vomiting [21, 22]. While SMV, is explored for its potential to enhance the quality of life in hepatocellular carcinoma patients [23, 24]. It has also been stated that CXB, DEX, and SMV may improve cancer treatment by targeting LD-induced chemoresistance based on their mechanisms of action. [19, 25, 26]. Likewise, a randomized controlled trial has shown significant reductions in cancer-related fatigue after DEX administration to patients with advanced cancers [27]. Since CXB, DEX, and SMV are used in some cancer treatment regimens to alleviate side effects caused by conventional chemotherapies, we explored their role in modulating the antiproliferative activity of 5-FU via an LD-dependent mechanism.
Interestingly, all tested drugs (CXB, DEX, or SMV) alone at their sub-toxic concentrations caused an elevation in LD formation [Figs. 4 (a-c)]. Our results regarding SMV-induced LD formation were in agreement with the findings of Gbelcová H et al. [28], as their study revealed that cell exposure to SMV led to enhance LD formation by altering cholesterol biosynthesis and increasing neutral lipid storage leading to an accumulation of cytosolic LDs in malignant and non-malignant cells. When administered in combination with 5-FU, SMV caused a decrease in the antiproliferative effects of 5-FU in MDA-MB-468 but not in A549 and HCT116 cells (Fig. 4a). Thus again, LD levels fail to serve as a predictive tool of response. It has been reported that SMV could potentiate the anticancer effects of 5-FU on bile duct cancer, which led to suggesting the use of SMV in regimens for managing bile duct cancer [29]. Moreover, strong antitumor activity in C26 colon carcinoma in vivo was shown after 5-FU/SMV treatment, suggesting that SMV acted as a sensitizer for tumor cells to 5-FU [30]. The difference in cellular response after the 5-FU/SMV combination indicates differences in pathways among various cancer origins. Increased intracellular LD levels in cancers have been reported to increase the incidence of 5-FU resistance [12, 31]. LDs contain many biologically active components, including sterol esters, TAGs, and functional proteins, which may all contribute to resistance but in different pathways that vary among different cancers [5]. Therefore, it might be assumed that LDs would induce modulation of growth/apoptosis through multiple pathways [7, 8]. Some of these pathways may be critical in some cancer types but not in others. SMV inhibits cholesterol synthesis by inhibiting HMG-CoA reductase enzyme. Interrupting such pathways may result in an imbalance in growth/death signals, leading to changes in cellular response behavior. To employ SMV in palliative regimens, a comprehensive understanding of LD-mediated chemoresistance pathways is needed, taking into account the differences in cancer origins.
Despite the increase in LD levels in all cells exposed to the 5-FU and CXB combination, CXB caused a reduction in 5-FU antiproliferative effects in HCT116 and MDA-MB-468, but not in A549 cells (Fig. 4a–c). Previous studies have shown comparable findings regarding the influence of CXB on 5-FU-induced anticancer activity. For instance, Lim et al. found that CXB attenuated the cytotoxic effect of 5-FU in HCT-15 and HT-29 human colon cancer cells [32]. They estimated that this decrease in 5-FU anticancer activity is mediated via changing the pattern of the cell cycle, leading to reduced apoptosis and 5-FU-induced accumulation of cells at sub-G1, which in turn inhibits the cell cycle-dependent anticancer activity of 5-FU. In contrast, Zhang et al. found that CXB combined with 5-FU enhanced apoptosis and antitumor efficacy in a subcutaneous implantation tumor model of human colon cancer [33]. Similar to the other tested palliative drugs, DEX induced a further increase in LD levels in HCT116, MDA-MB-468, and A549 cells. Likewise, the addition of DEX antagonized the antiproliferative effects of 5-FU only in the former two cell lines and not in A549 (Fig. 4 c). Carollo et al. demonstrated a diminished cytotoxic activity and increased resistance to 5-FU in PC-3 cells when combined with DEX [34]. According to those researchers, this drop in 5-FU activity might be mediated by the induction of the anti-inflammatory protein lipocortin-1 and the inhibition of arachidonic acid metabolism. These findings also indicate that the combination of DEX with conventional chemotherapeutic agents may result in an antagonistic effect. Additionally, DEX was shown to stimulate de novo TAG synthesis according to Dolinsky et al. [35]. Li et al. supported the notion that activation of TAG biosynthesis protects cells from lipid peroxide-induced membrane damage under increased levels of oxidative stress during apoptosis [36]. They suggested that targeting TAG biosynthesis in cancer cells might represent a new approach to promoting cell death during apoptosis.
The study demonstrates that the co-administration of DEX, CXB, and SMV with 5-FU may induce chemoresistance in certain cancer cell lines through LD accumulation, potentially diminishing therapeutic efficacy of 5-FU. This chemoprotective effect was evident in MDA-MB-468 and HCT116 cells, but absent in A549 cells, suggesting a cancer-specific LD-mediated resistance mechanism. If this resistance is confirmed in clinical settings, adjustments in treatment protocols could be necessary to maintain chemotherapeutic outcomes. Further research is essential to elucidate this pathway and identify potential targets to counteract chemoresistance, offering avenues for more tailored and effective cancer therapies.


Comments (0)