Bacterial strains, reagents, plasmids, and growth conditions
The microbiological media used in this study were purchased from Oxoid Ltd (Basingstoke, Hampshire, England) and Sangon Biotech (Shanghai, China); the reagents and antibiotics were purchased from Sigma-Aldrich (St. Louis, MO, USA). The V. parahaemolyticus SH112 strain (GenBank: JACYGZ000000000.1) was isolated from a clinical specimen in Shanghai and stocked in the China General Microbiological Culture Collection Center under the accession number CGMCC 1.90013 and was used as the WT strain (Li et al. 2022). The V. parahaemolyticus strains in this study were cultured in Luria–Bertani (LB) broth supplemented with 2% sodium chloride at 30 °C, with shaking at 160 rpm (Noh et al. 2015). V. parahaemolyticus was identified using Thiosulfate-citrate-bile salts-sucrose agar culture medium (TCBS Agar; HKM, Guangdong, China). For gene cloning and the construction of gene deletion mutants, Escherichia coli CC118 λpir was grown in LB medium at 37 °C (Li et al. 2022). The suicide plasmid pYAK1 and the plasmid pMMB207 were used to generate gene deletion mutants and complemented strains (Li et al. 2022). The WT and derivative strains of V. parahaemolyticus and E. coli are listed in Table S1 of the supplemental material. Antibiotics were administered at the following concentrations: 10 μg/mL chloramphenicol and 70 μg/mL kanamycin.
Bacterial concentrations in LB medium were estimated using 96-well microtiter plates (100 μL) based on absorbance OD600 (optical density at 600 nm) values recorded with a microplate spectrophotometer (Multiskan GO, Thermo Fisher Scientific, Waltham, MA, USA), or using the standard plate count method with incubation at 30 °C for 16 h in LB medium containing 1.5% agar.
For swimming motility and transmission electron microscopy (TEM) assays, culture plates containing LB medium with 0.3% agar (0.3% agar-LB plates) were incubated at 30 °C overnight. For swarming motility assays, heart infusion (HI) medium plates containing 1.5% agar (1.5% agar-HI plates) were used. For colony morphology assays, LB medium plates containing 2% agar (2% agar-LB plates) and Congo red (CR) plates were used. The CR plates were prepared using 2% agar-LB after autoclaving and then by adding 40 μg/mL filtered Congo red and 20 μg/mL Coomassie brilliant blue R (Yuanye Bio-Technology, Shanghai, China).
Construction of deletion mutants
We followed previously published methods (Yu et al. 2012; 2015) to generate the vpA1040 deletion mutant strain. Briefly, two DNA fragments were amplified by PCR, using WT DNA as the template and the primer pair tssL2-A/B or tssL2-C/D, respectively (Table S2 in the supplemental material). Next, the tssL2-AB (425 bp upstream of the start codon) and tssL2-CD (487 bp upstream of the stop codon) fragments were used as the templates with the primers tssL2-A/D to construct a tssL2 deletion fragment via PCR amplification. After recovery and purification using agarose gel, the deletion fragment was digested with restriction enzymes BamHI and SphI, ligated into pYAK, and then digested with the same restriction enzymes to produce pYAK-tssL2-AD. This plasmid was transformed into E. coli CC118 λpir to obtain the strain pYAK-tssL2-CC118 λpir, which was then conjugated into the V. parahaemolyticus SH112 WT strain. TCBS agar plates supplemented with chloramphenicol (10 μg/mL) and LB agar plates containing 20% sucrose were used to screen the V. parahaemolyticus cells with the gene deletion from one generation to another until clones were unable to grow on the TCBS plates. After continuous culturing, suspected positive clones were isolated, cultured in LB, and confirmed by PCR with tssL2-E/F primers (deletion mutants, 1222 bp). Meanwhile, sacB-E/F primers (deletion mutants, 500 bp) were used to confirm whether the deletion mutant had discarded the pYAK plasmid. After further confirmation by sequencing, positive colonies were designated as the ∆tssL2 strain.
Construction of complementary strains
The entire open reading frame fragment of tssL2 (1538 bp) was amplified by PCR with V. parahaemolyticus SH112 DNA as the template and tssL2-pMMB-F/R primers (Table S2 in the supplemental material). After digestion with restriction enzymes EcoRI and BamHI, the sequence was ligated to the digested pMMB plasmid to obtain the plasmid tssL2-pMMB. This plasmid was transformed into and propagated in E. coli CC118 λpir. Then, tssL2-pMMB-CC118 λpir was conjugated to ∆tssL2, which further developed into a complementary strain through several generations under chloramphenicol selection. The positive complementary strains were identified using PCR with tssL2-pMMB-F/R primers and were ultimately designated as C∆tssL2.
Sequence analysis of tssL2 gene and TssL2 protein
The nucleotide sequences of tssL2 gene from other V. parahaemolyticus strains and representative vibro spp. were downloaded from NCBI and aligned with the sequence of TssL2 in this study using Mesquite software (Zhang et al. 2020). In addition, the TssL2 protein structure of Vibrio paraholyticus was analyzed by protein structure online prediction website (Song et al. 2020).
Extraction of mRNA and qRT-PCR
QRT-PCR was used to assess the transcriptional level of corresponding mRNAs in V. parahaemolyticus strains and infected cells, as reported previously (Yu et al. 2012). Total RNA was isolated from centrifuged bacteria or collected cells using the TRizol® reagent (Invitrogen, Carlsbad, CA, USA). Residual DNA digestion and cDNA preparation were performed using a Prime Script® RT reagent Kit with gDNA Eraser (RR047A, Takara Bio, Shiga, Japan) according to the manufacturer’s protocol. Real-time PCR was performed on the Applied Biosystems® 7500 Fast Real-time PCR system (4,406,985, Applied Biosystems, Waltham, MA, USA) using SYBR® Premix ExTaq™ П (RR820A, Takara Bio, Shiga, Japan) and designed gene-specific primers (see Table S3 in the supplemental material) in a reaction mixture with a total volume of 20 μL. The PCR thermocycling protocol comprised 30 cycles of 94 °C for 15 s, 54 °C for 30 s, and 68 °C for 1 min. Using the comparative cycle threshold (2−∆∆CT) method (Wang et al. 2016), relative gene expression was normalized to the expression of the housekeeping gene gap in V. parahaemolyticus. Experiments were repeated with three biological and three technical replicates.
Analysis of transcription, expression and secretion of T6SS2
For analysis of T6SS2 transcription, the genes vgrG2 (vpA1026), hcp2 (vpA1027), tssM2 (vpA1039), tssL2 (vpA1040), tssK2 (vpA1041), vpA1044 were selected for detection using qRT-PCR (see Table S3 in the supplemental material). Three strains (WT, ∆tssL2, and C∆tssL2) were harvested in LB medium and then grown until reaching the logarithmic growth phase (OD600 = 0.2), and extraction of mRNA and qRT-PCR were performed.
For analysis of T6SS2 expression and secretion, Hcp2 was selected for detection using Western blot (Salomon et al. 2013). High-density V. parahaemolyticus strains were acquired by culturing in LB medium at 28 °C for 4 h. The strains were then cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco®, Grand Island, NY, USA) with low-glucose and low-salt conditions for 6 h at 28 °C and centrifuged at 5000 × g for 30 min. The secreted supernatant proteins and pellet proteins were collected and separated using 12% sodium dodecyl sulfate–polyacrylamide gels. The proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences, Amersham, UK) using the Trans-Blot® SD semi-dry transfer cell (Bio-Rad, USA). Rabbit anti-thermostable direct hemolysin (TDH) antibody and anti-cytosolic marker AMP receptor protein (Crp) antibody were used as a positive and lysis control, and anti-Hcp2 polyclonal antibodies were used to specifically bind to secreted proteins, which were then detected with goat anti-rabbit IgG-HRP (Sigma-Aldrich, St. Louis, MO, USA).
Bacterial killing assays
As previously described (Salomon et al. 2014), V. parahaemolyticus strains were incubated overnight, subcultured in fresh LB medium and then grown until reaching the logarithmic phase (OD600 = 0.2). After washing and resuspending in 1 × phosphate-buffered saline (PBS), the three V. parahaemolyticus strains were mixed with E. coli DH5α in a 4:1 ratio and analyzed in triplicate. Next, the bacterial mixture was split into two samples; one was incubated at 30 °C for 4 h, and the other was used to spot tenfold serial dilutions on LB and LB plus ampicillin plates to determine initial concentrations of E. coli and V. parahaemolyticus. The V. parahaemolyticus strains used in this study were resistant to ampicillin, as recently reported (Mala et al. 2016), but DH5α was not. After 4 h, incubated suspensions were cultured on LB and LB with ampicillin plates to confirm the bacterial survival rate; these assays were repeated three times.
Aggregation assay
As reported previously (Levinson et al. 2015; Liu et al. 2012), overnight cultures of all strains were diluted to 1:100 with 5 mL of LB medium in glass test tubes and grown to equal OD600 values (approximately 0.25). All the tubes were shaken vigorously for 15 s before the start of the assay and were then statically cultured at 22–24 °C for 72 h. Four replicates were prepared for each strain. One of the tubes was used to photograph the culture suspension, and the other three replicates were used to regularly confirm OD600 values of 100 μL samples from the liquid surface to determine aggregation. The experiment was performed independently in triplicate.
Evaluation of bacterial biofilms
Following a published crystal violet staining method (Kimbrough et al. 2020), V. parahaemolyticus strains were subcultured into fresh LB and grown to the logarithmic phase (OD600 = 0.2) by shaking at 30 °C for 6 h. The strains were diluted to 1:100 in 5 mL LB in glass test tubes and were grown at room temperature with no shaking for 72 h. Biofilms at the air–liquid interface were visualized and imaged. Notably, as the newly formed biofilm was highly vulnerable to destruction, the tube should not be moved during the process of biofilm formation, and extra care must be taken when moving the tube for imaging. Then, cultures were carefully decanted to acquire the biofilm and associated cells adhering to the walls of the tube. After the biofilm was air-dried, it was stained with 5 mL of 1% Crystal Violet for 30 min at 22–24 °C and then thoroughly washed with 1 × PBS. Finally, the adherent biofilm was solubilized using 5 mL of 100% ethanol for 10 min, then the absorbance was measured at 600 nm. The experiment was performed at least three times.
In addition, qRT-PCR for biofilm-related regulatory genes (calR, cspQ, mfpA, aphA, opaR, luxS, mshA, oxyR, cspA) in V. parahaemolyticus SH112 were performed (Faleye et al. 2021). For qRT-PCR, the strains were grown in 2.5% (w/v) HI broth (ELITE Biotech, Shanghai, China) at 37 °C shaking at 250 rpm. Briefly, overnight cultures were 50-fold diluted in 15 mL of fresh HI broth, and harvested at the mid-exponential growth phase (OD600 = 0.5). Subsequently, resulting cultures were diluted 1000-fold into 15 mL of fresh HI broth for a third round of cultivation, and extraction of mRNA were performed as described above. When necessary, 50 μg/mL gentamicin was supplemented in the medium.
Colony morphology
The colony morphology assays were performed as described (Kimbrough et al. 2020; Martín-Rodríguez et al. 2021), with modifications. Briefly, the strains were statically cultured in LB medium until reaching the logarithmic growth period. Next, 2.5 μL samples of the resultant culture were inoculated on 2% agar-LB plates and CR plates and incubated at 30 °C for 48 h before culturing for 5 days at 22–24 °C. All colony morphology images were obtained on day 6.
Motility assay
As described previously (Guo et al. 2019), the strains were freshly grown in LB broth with 2% sodium chloride at 30 °C until they reached equal optical densities (OD600 = 0.2). To observe swimming motility, 2 μL of each culture sample was inoculated on the surface of 0.3% agar-LB plates with 2% sodium chloride and incubated for 4 h at 30 °C. For the swarming assay, cultures were performed similarly to the swimming assays but on 1.5% agar-HI plates for 24 h at 30 °C. For both motility assays, the diameters of the bacterial motility halos were measured and recorded.
Observation and gene transcription assays of flagella
The ultrastructures of the flagella of the WT and ∆tssL2 strains were observed through high-resolution TEM (FEI, Ltd., Hillsboro, OR, USA) using the negative staining method with phosphotungstic acid to examine correlation of morphological factors with motility (Li et al. 2022). The strains were separately inoculated on 0.3% agar-LB plates with 2% sodium chloride and incubated for 4 h at 30 °C to observe bacterial polar flagella (Noh et al. 2015). Single colonies of each strain were then gently suspended in 1 × PBS and negatively stained with 2% phosphotungstic acid (Sigma-Aldrich, St. Louis, MO, USA). The stained bacteria were adhered to grids, air-dried, and observed with TEM. The ratios of damaged and fractured polar flagella (including surrounding or scattering attached flagella) in three strains were calculated as the number of the polar flagella damaged and fractured/the total number of the polar flagella × 100%. To validate the TEM results, qRT-PCR was used to measure the transcription levels of the flagellar protein-associated genes of V. parahaemolyticus on chromosomes I and II in polar and lateral flagella (Masum et al. 2017; Noh et al. 2015). To obtain swimming- and swarming-related mRNAs, the strains were assembled on 0.3% agar-LB plates with 2% sodium chloride for 4 h post-incubation and on 1.5% agar-HI plates for 24 h. The structural polar flagellar (flaA, flgE, flgM, fliD, fliF, fliG, motB) and lateral flagellar (lafA, flgB, flgM, fliD, fliE, fliM, motB, motY) genes were selected for detection using qRT-PCR (see Table S3 in the supplemental material). The experiments were repeated in triplicate, with four replicates per strain.
Adhesion and cytotoxicity assay
Adherence and cytotoxicity tests of V. parahaemolyticus strains on Caco-2 cell monolayers were performed using as previously described (Yu et al. 2012; Ming and Sheng 2015). Briefly, Caco-2 cells were grown in DMEM supplemented with 20% FBS without antibiotics. For adherence assay, monolayer cells (approximately 5 × 105 cells per well in 24-well cell plates) were co-cultured with PBS-washed bacteria in DMEM at a multiplicity of infection (MOI) of 1:10, and incubated at 37 °C for 1 h. After washed twice with 1 × PBS, the cells completely lysed with 0.5% Triton X®-100. Subsequently, serial tenfold dilutions of bacteria-containing lysates were plated onto LB agar plates, and the number of single colonies of each strain was recorded. Percent adherence was expressed as bacterial cells adhered/bacterial cells added into the well × 100.
For cytotoxicity assay, Caco-2 cells were cultured in a 96-well plate (approximately 2 × 104 cells per well). Before infection, the cell monolayers were washed with 1 × PBS and were inoculated with three strains at an MOI of 1:10 in DMEM, then respectively incubated at 37 °C for 0.5, 1, 1.5, 2, 2.5, 3 h. The culture supernatants containing lactate dehydrogenase (LDH) were collected according to the manufacturer’s instructions for the CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit (Promega, Madison, WI, USA). The spontaneous (positive) and maximum (control) release of LDH was measured from the cytoplasm of uninfected Caco-2 cells without or with the addition of 0.8% Triton X-100, respectively. Sample absorbance values (OD490) were measured using a microplate reader and expressed as the percentage of the positive and control values. Percentage cytotoxicity was calculated using the formula: (test LDH release − spontaneous release)/maximal release.
Acute toxicity studies in mice
As previously described (Flood and Kondo 2004), acute toxicity testing was performed on 4-week-old, specific-pathogen-free ICR (Institute of Cancer Research) mice purchased from the Slack Shanghai Laboratory Animal Co., Ltd. (Shanghai, China). The mice were randomly assigned to 15 treatment groups, each containing eight mice. When growth reached the logarithmic phase (OD600 = 0.2), bacterial suspensions were adjusted to 200 μL doses per mouse of 4 × 107, 2 × 107, 1 × 107, 5 × 106, and 2.5 × 106 CFU. The mice were injected intraperitoneally with different bacterial concentrations for the three strains. In addition, five mice were inoculated with sterile physiological saline as the control. The animals were observed for 7 days, and 50% lethal dose (LD50) was calculated using the Reed–Muench method (Yi et al. 2013).
Infection experiments in mice
After confirming the LD50 of an intermediate dose (1.5 × 107 CFU per animal), four groups containing twelve 4-week-old ICR mice each received an intraperitoneal injection of bacteria (WT, ∆tssL2, or C∆tssL2) or sterile physiological saline. Survival was monitored once every 5 h until 7 days post-infection. Three mice were selected from each group at 20 h post-infection to assess the presence of viable bacteria in infected organs. Blood samples were drawn by ocular enucleation, and an anticoagulant (sodium citrate) was added. After aseptic anatomical sampling, tissue (0.2 g and 0.1 g from the liver and spleen, respectively) and blood samples (100 μL) were homogenized, diluted tenfold in sterile physiological saline, and plated onto TCBS agar to evaluate bacterial colonization. By counting the bacterial colonies on the plates, the degree of bacterial colonization in the blood, liver, and spleen of ICR mice was calculated.
Cytokine assays
Caco-2 cells in 24-well plates were infected with the three strains at an MOI of 1:100, with uninfected Caco-2 cells in the medium serving as the control group. After infection with the V. parahaemolyticus strains for 1 h, monolayer Caco-2 cells were treated with gentamicin (200 μg/mL), then washed three times with 1 × PBS. After culturing for 1, 2, 3, and 4 h, Caco-2 cells were collected, and total mRNA was extracted for RT-PCR (Zhou et al. 2013a, b). The primer sequences for interleukin-6 (IL-6), IL-8, and β-actin (as an internal control) are listed in supplemental material Table S3.
Sample preparation and label-free quantitative mass spectrometry
Three biological replicates of the WT and ∆tssL2 strains (WT-1, WT-2, WT-3 and ∆tssL2-1, ∆tssL2-2, ∆tssL2-3) were grown in 50 mL DMEM media at 28 °C with vigorous agitation for 6 h, and centrifuged at 5000 × g for 30 min at 4 °C. The secretory proteins in the culture supernatant were prepared using a customized procedure by label-free quantitative mass spectrometry (APTBIO, Shanghai, China). Briefly, secretory proteins in 40 mL of culture supernatant were freeze-dried by liquid helium and cleaved with 1 mL SDT (%[w/v] SDS, 100 mM Tris/HCl pH 7.6, 0.1 M DTT), then quantified using the BCA protein assay. Collected proteomes were lysed with trypsin to complex peptide mixtures by filter-aided sample preparation, and then the peptides were desalted by C18 cartridge. Each sample was separated by high performance liquid chromatography using the EASY-nLC1200 system, and the separated peptides were eluted with water containing 0.1% formic acid (FA) and 84% acetonitrile containing 0.1% FA at a flow rate of 300 nL min−1. Peptide analysis was performed with Q-exactive mass spectrometer, and the differentially expressed proteins (DEPs) were subjected to bioinformatics analysis in accordance with the criteria of p value of < 0.05 and fold changes > 2.0 or < 0.5. GO and KEGG enrichment analyses based on Fisher’s exact test were performed.
Gene transcription assays of T3SS1 and T2SS
QRT-PCR was used to measure the transcription levels of T3SS1 and T2SS genes of V. parahaemolyticus (Lian et al. 2022). To obtain mRNAs, the strains were cultured in LB medium at 37 °C until they reached the logarithmic phase (OD600 = 0.2). The T3SS1 (vopD, vopB, vscP, vecA, vopR, vopS, vscJ, vscG) and T2SS (gspC, gspE, gspJ, gspL, gspM) genes were selected for detection using qRT-PCR (see Table S3 in the supplemental material).
Statistical analysis
The TEM observations were repeated twice, and other assays were repeated thrice, each with least three replicates per strain. Data were displayed as mean ± standard deviation (SD) and analyzed in Graph Pad Prism (v5; Systat Software Inc., Chicago, IL, USA). Differences were considered significant when P < 0.05, as denoted by one asterisk (*); outstanding significance was achieved when P < 0.01, as denoted by two asterisks (**); and extremely remarkable significance was achieved when P < 0.001, as denoted by three asterisks (***).

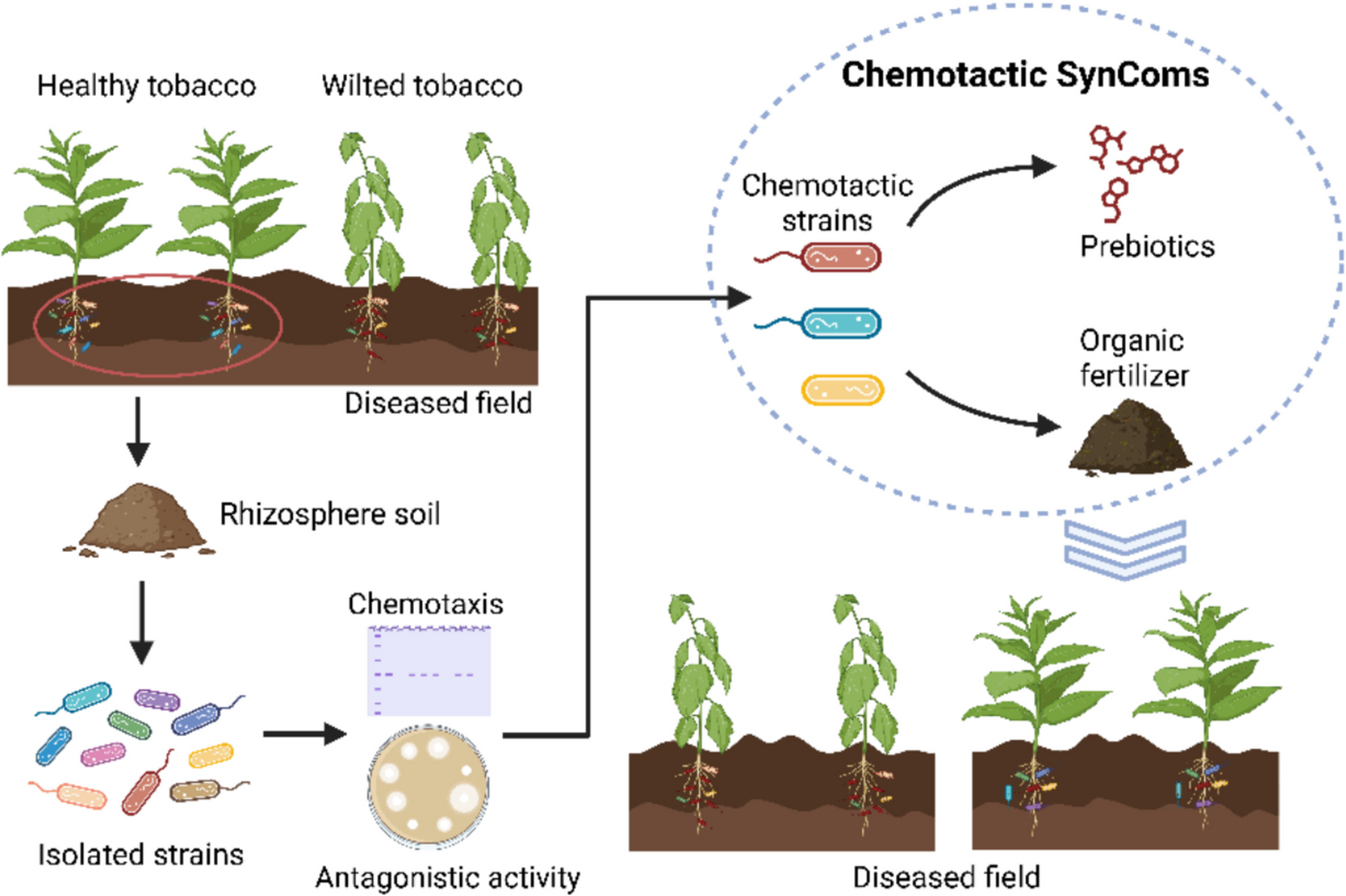
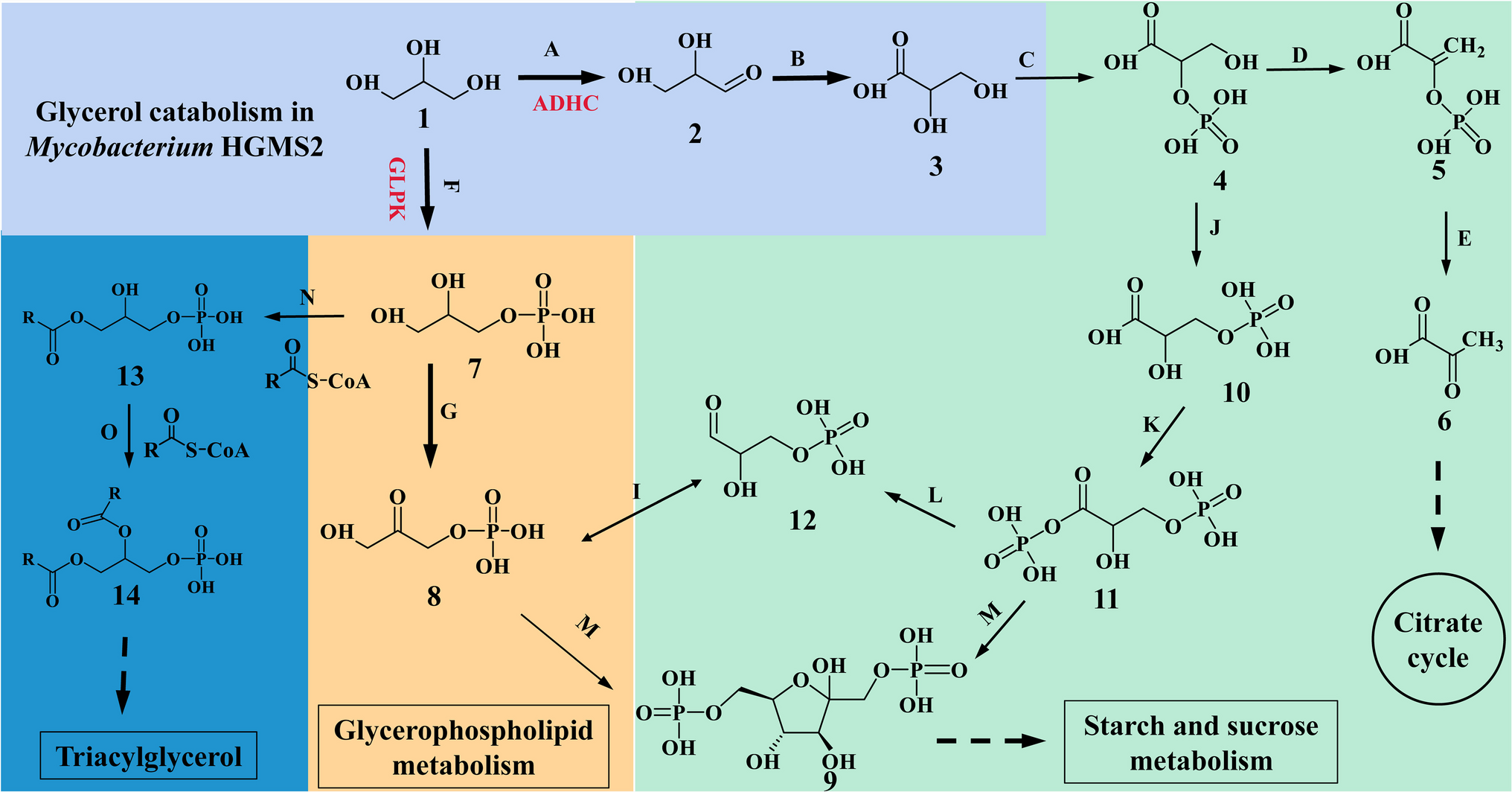
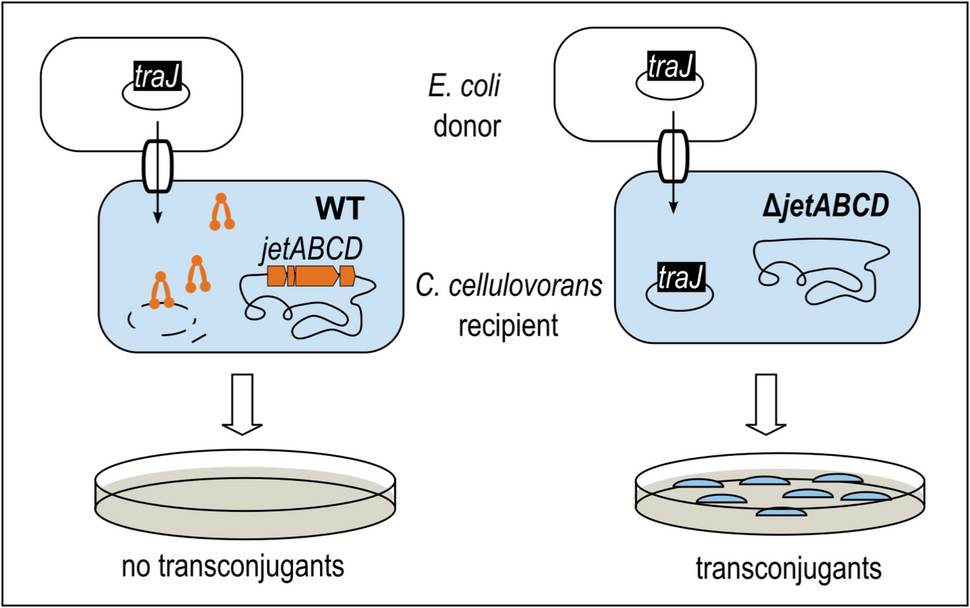
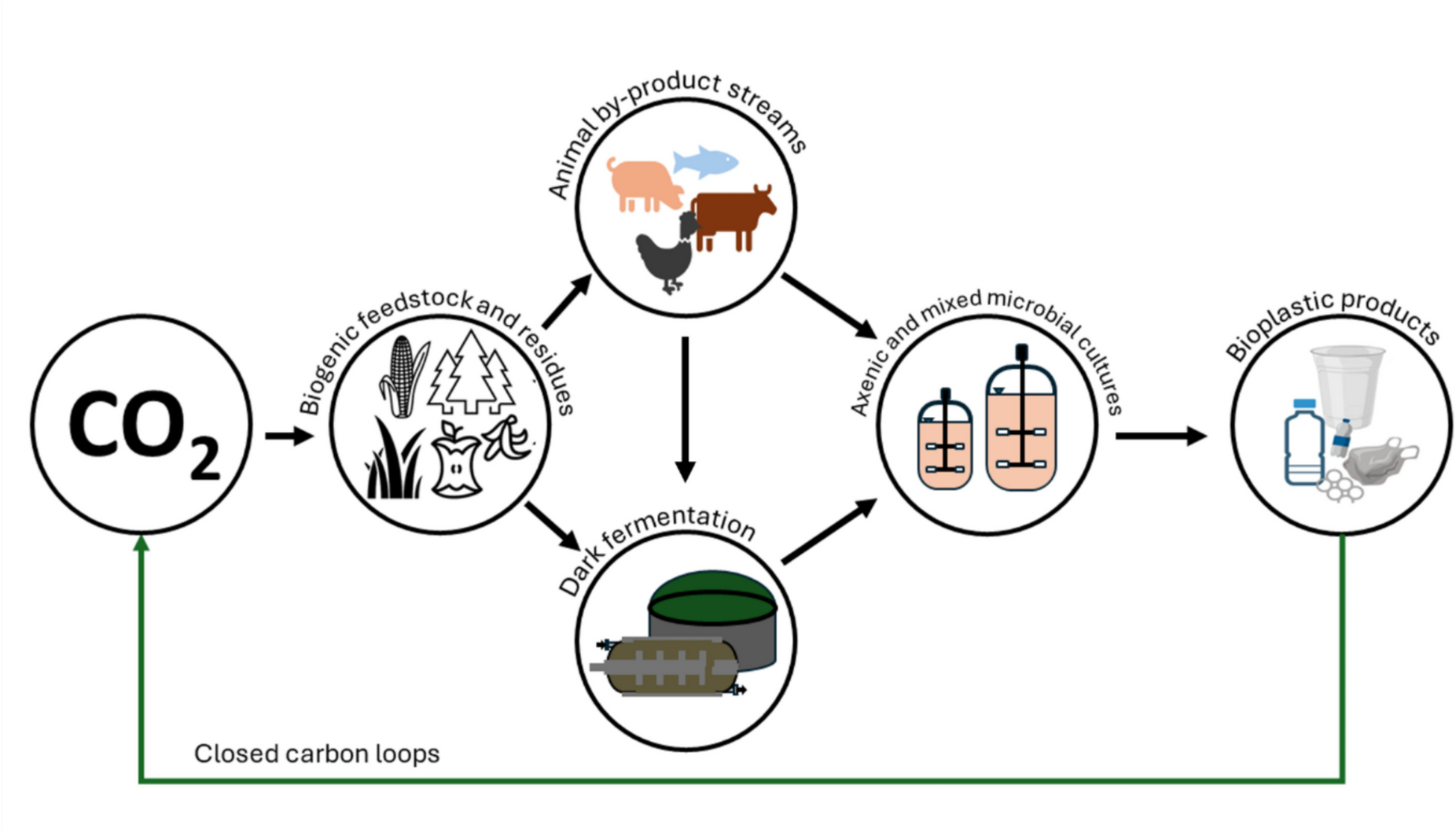
Comments (0)