
Remember me



For a long time, the concept of neurodegeneration centered essentially on neuronal soma death [8]. However, this view has since broadened to encompass synaptic dysfunction and disruption of neuronal connectivity [9]. In the context of PD, it is important to highlight key characteristics of SNpc axonal biology. First, neurons in this region possess long, unmyelinated axons, with extensive branching, allowing them to form numerous synapses with medium spiny neurons in the striatum. It is estimated that a single SNpc neuron can establish connections with around one million striatal neurons [2]. Due to this unique morphological feature, these neurons exhibit high bioenergetic demand [8]. The combination of elevated mitochondrial activity and dopamine metabolism at axonal terminals creates an environment prone to high oxidative stress, rendering these neurons particularly vulnerable to degeneration [2]. Another significant aspect to consider is the role of α-syn accumulation. Under physiological conditions, this protein is involved in synaptic vesicle recycling. Notwithstanding, in PD, it misfolds and accumulates within axonal terminals, disrupting synaptic function and leading to an increase in cellular oxidation through its interaction with DA, ultimately contributing to progressive neuronal degeneration [8].
Compelling evidence indicates that α-syn accumulation, a key pathological feature in PD, begins mainly in the axonal terminals, triggering a retrograde degenerative process toward the soma. Thus, when neuronal death begins to occur in the SNpc, nigral projections in the striatum may already be severely compromised [6]. Consistently, early co-accumulation of α-syn and synapsin III at synaptic terminals has been observed in the initial stages in a MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced mouse model of parkinsonism, thereby contributing to nigrostriatal denervation [10]. The same study also reported a notable reduction in the immunoreactivity of key proteins involved in dopaminergic signaling at early stages, specifically the DA transporter (DAT) and vesicular monoamine transporter 2 (VMAT2). These proteins play crucial roles in regulating DA levels within the striatum.
Strikingly, although VMAT2 is primarily recognized for its role in vesicular DA storage, it also contributes significantly to the packaging of gamma-aminobutyric acid (GABA) within the nigrostriatal pathway [11]. This suggests co-release of GABA and DA from SNpc projections to the striatum, as GABA release in the nigrostriatal terminals does not rely on the vesicular GABA transporter (VGAT), highlighting the importance of VMAT2 [12]. GABA release by SNpc dopaminergic neurons appears to play a crucial role for striatal neurons functioning and basal ganglia circuitry [12]. Moreover, it is involved in the autoregulation of phasic DA release through its binding to GABAA receptors located on DA axons within the striatum. Notably, SNpc DA axons lack the molecular machinery required for GABA synthesis; instead, most GABA is taken up from neighboring striatal cells [13]. Taken together, these results underscore the pivotal role of processes carried out at DA SNpc axon terminals in modulating striatal and basal ganglia activity. Nevertheless, the mechanisms and implications of GABA/DA co-release in PD remain to be fully elucidated.
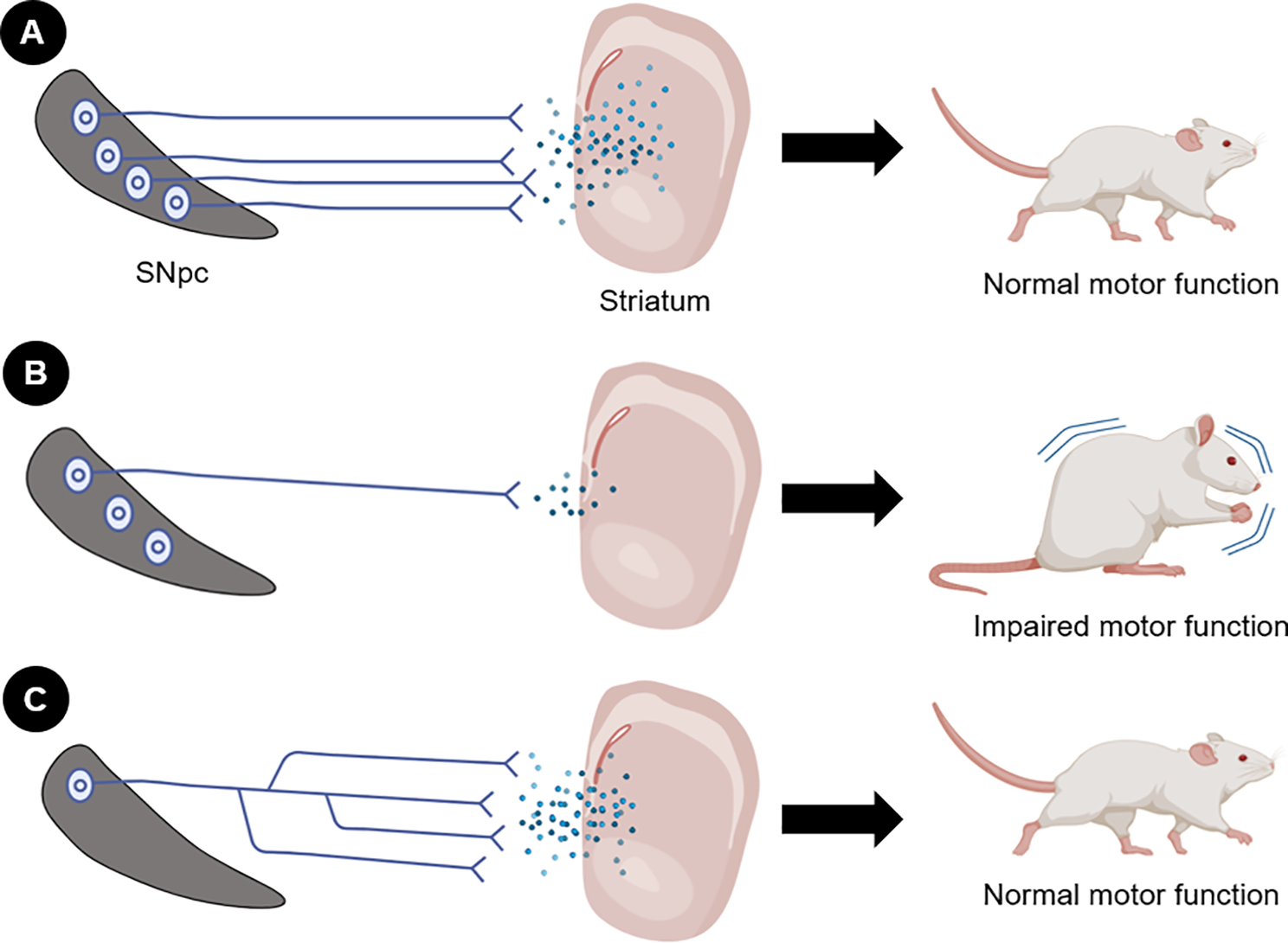
A recent study further supports for the role of axonal degeneration in the SNpc in the emergence of motor symptoms in PD [7]. The research focused on the impact of a mitochondrial DNA mutation–K320E-TwinkleDaN – on dopaminergic neurons. This mutation significantly accelerates the processes involved in neuronal degeneration. Remarkably, after 20 months, mutant mice exhibited normal motor function, despite the loss of ∼70% of nigral dopaminergic neurons. The remaining neurons maintained ∼75% of axon terminals in the dorsal striatum, preserving normal neurotransmission. This maintenance of motor function was attributed to compensatory axonal sprouting from surviving neurons, which sustained striatal innervation (Fig. 1). Enhanced axon sprouting from the SNpc was associated with increased levels of unconventional neurotrophic factors, particularly netrin 1 (Ntn1) and ephrin-A2 (Efna2). In contrast, there were reduced levels of semaphorin 3 A (Sema3A) and Slit2, which are known to inhibit axon branching.
It is worth highlighting that in PD, α-syn pathology extends beyond the nigrostriatal pathway. Early accumulation of α-syn can occur in peripheral tissues and may spread in a “prion-like” manner from cell to cell via axons and synapses across neural circuits [13]. Recent studies have shown that pathological α-syn accumulates in the gut, liver [13], spinal cord, and kidneys [14] before overt neuronal damage occurs, suggesting early peripheral involvement. Alternatively, α-syn pathology may remain largely confined to the CNS, reflecting variability in both disease onset and progression.
Fig. 1
Role of nigrostriatal axon integrity for the maintenance of normal motor function. (A) Under physiological conditions, neurons and axons are preserved, sustaining neurotransmission and normal motor function. (B) Under pathological conditions such as PD, there is loss of cell bodies and, more notably, axonal degeneration, leading to reduced nigrostriatal neurotransmission and consequent motor impairment. (C) Surviving neurons in the substantia nigra undertake a compensatory mechanism to maintain striatal innervation. In this case, axonal sprouting occurs, preserving nigrostriatal neurotransmission and motor function
Comments (0)