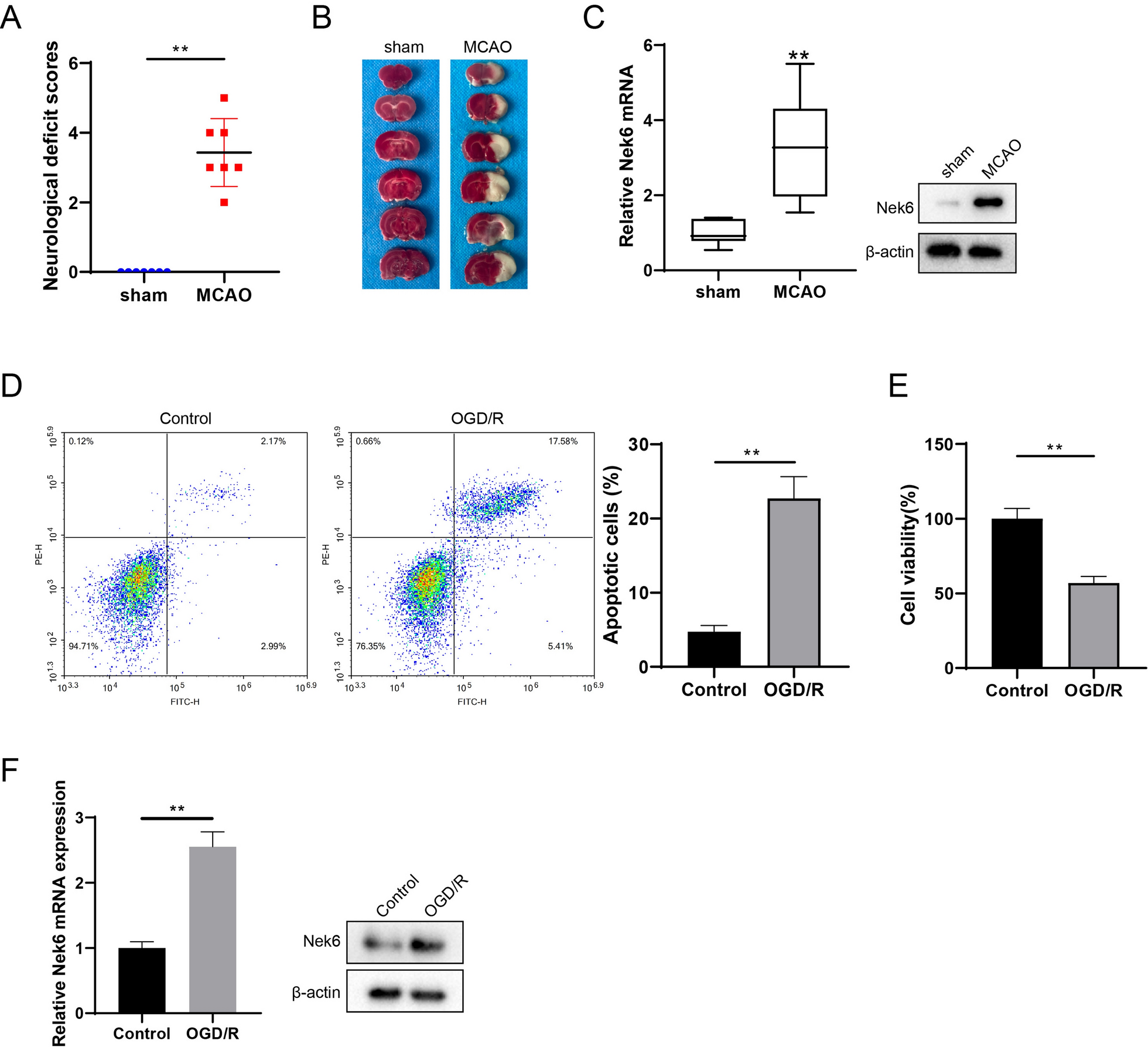
Establishment of CIRI mice model by MCAO
All animal experiments are approved by the Ethics committee of The First Affiliated Hospital of Zhengzhou University. C57BL/6 J male mice (8–10 weeks, weight 23–25 g, Zhejiang Vital River Laboratory Animal Technology Co., Ltd.) were fasted for 12 h before surgery, and the MCAO model was established after anesthesia [13]. A 6-0 single-strand nylon wire was inserted from the bifurcation of the external carotid artery (ECA) or common carotid artery (CCA) into the internal carotid artery (ICA). The internal carotid artery was inserted to block the beginning of the middle cerebral artery (MCA) and all its blood supply, resulting in focal ischemia in the MCA supply area. After 1 h ischemia, the thread plug was withdrawn slowly. Only the ECA, CCA, ICA and MCA were separated, and the group without wire insertion was used as the sham group. Brain tissues from one mouse in the sham group (n = 7) and MCAO group (n = 7) was randomly selected for 2,3,5-triphenyltetrazolium chloride (TTC) staining, and single-cell suspension of peri-infarct brain tissues from the remaining 6 were taken for subsequent experiments.
Intracerebroventricular injection
After MCAO establishment, the mice received one microinjection with AAV-Nek6 or AAV-NC (1 μL of 1 × 109 viral genomes/μL) in the ipsilateral cortex ischemic lesions, and then grouped into MCAO + AAV-NC (n = 10), MCAO + AAV-Nek6 (n = 10). The coordinates of the microinjection were the following: 0.3 mm anterior to the bregma, 3 mm lateral, 2 mm deep and 1.9 mm posterior to the bregma, 3 mm lateral, 2 mm deep. The injection was completed with a 10 μL microsyringe at the rate of 0.1 μL/min, and the microinjector was placed for 10 min after injection. Brain tissues from one mouse in the MCAO + AAV-NC and MCAO + AAV-Nek6 group was randomly selected for TTC staining, and peri-infarct brain tissues from the remaining 4 were taken for subsequent experiments.
Neurological deficit scores and TTC staining
Each group of mice was scored according to the modified neurological severity scores (mNSS). After scoring the neurological severity, the brains were removed by anesthesia and frozen at -20 °C for 20 min, then cut into 2 mm serial coronal sections in a brain tank and incubated with 0.2% TTC staining solution (Solarbio) for 30 min at 37 °C in a light-protected environment. The infarcted areas were pale white and the normal brain tissue was dark red after staining.
TUNEL assay
Brain tissues from each group were dehydrated, embedded, sectioned and incubated in 0.9% NaCl injection for 5 min. Then the slices were incubated in TdT reaction mixture for 60 min at 37 °C. After washing with PBS, the tissues were blocked with 0.3% H2O2, 50 μL of TUNEL reaction mixture (Roche) was added, and incubated in the dark for 60 min at 37 °C in a humid environment. The sections were washed three times. The number of TUNEL-positive cells was observed under fluorescence microscope and photographed.
Quantitative real-time polymerase chain reaction (QPCR)
Total RNA was extracted from single-cell suspension of peri-infarct brain tissues or SH-SY5Y cells using miR-Neasy Mini Kit (QIAGEN China (Shanghai) Co., Ltd.) and reverse transcribed into cDNA according to Taq Man MicroRNA Assays Reverse Transcription Primer instructions (QIAGEN China (Shanghai) Co., Ltd.). QPCR was performed according to SYBR Green PCR kit (Shanghai solarbio Bioscience Technology Co., LTD) instructions. β-actin was used as an internal reference and calculated using the 2−△△Ct method.
Western blot
Total protein from single-cell suspension of peri-infarct brain tissues or SH-SY5Y cells was extracted using RIPA lysis buffer (Beyotime), and protein concentrations were quantified by BCA Protein Assay Kit (Beyotime) according to the manufacturer's instructions. Proteins were separated by 8–10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After being closed in 5% skim milk powder for 1 h at room temperature, the membranes were incubated with anti-Nek6 (santa cruz, sc-374491), Beclin 1 (Abcam, ab207612), autophagy marker light chain 3B (LC3B, Abcam, ab221794), phospho-Akt (p-Akt, Ser473, cell signaling technology, #4060), Akt (cell signaling technology, #4685), phospho-mTOR (p-mTOR, Ser2448, cell signaling technology, #5536), mTOR (cell signaling technology, #2983), adenosine 5’-monophosphate-activated protein kinase α (AMPKα, cell signaling technology, #5831), p-AMPKα (Thr172, cell signaling technology, #50081), UNC-51-like kinase 1 (ULK1, Thermo Fisher Scientific, PA5-34542), p-ULK1 (Ser317, Thermo Fisher Scientific, PA5-104556), protein phosphatase 1α (PP1α, Thermo Fisher Scientific, 43-8100), p-PP1α (Thr320, Thermo Fisher Scientific, PA5-17819) at 4 °C overnight. The membranes were then incubated with secondary antibodies conjugated to HRP for 1 h at room temperature. The membranes were incubated for 1 h. The membranes were chromogenized using the EasyBlot ECL kit and the protein blot intensity was quantified by an Amersham Imager 600 system [14].
Cell culture and establishment of oxygen glucose deprivation/reoxygenation (OGD/R) model
The neuronal cell line SH-SY5Y was provided by the Shanghai Cell Bank of the Chinese Academy of Sciences and cultured in DMEM medium containing 10% fetal bovine serum in a 5% CO2 incubator at 37 °C. SH-SY5Y cells were co-deprived of O2 and glucose for 1 h to simulate the in vitro injury model, after removing the original medium and adding sugar-free medium DMEM, and placed in a hypoxic chamber in saturated gas with 95% N2 and 5% CO2, and the oxygen concentration was maintained within 1%, and the temperature was controlled at 37 °C.
Cell transfection and treatment
SH-SY5Y cells were inoculated in 6-well plates (1 × 105/well) and transfected with pcDNA and pcDNA-Nek6, si-NC and si-Nek6, si-NC and si-methyltransferase·like protein 3 (si-METTL3) according to Lipofectamine 2000 reagent when the cell fusion level reached 70%. 12 h after transfection, fresh DEME medium containing fetal bovine serum was replaced and the cells were continued to be cultured for 48 h. The cells were collected for subsequent experiments. In the OGD/R + pcDNA-Nek6 + LY or OGD/R + pcDNA-Nek6 + Rapa group, cells were treated with 50 mmol/L LY294002 (LY, AKT inhibitor) or Rapamycin (Rapa, mTOR inhibitor) for 1 h before transfection. In the OGD/R + pcDNA-Nek6 + RSVA405 group, cells were treated with 3 μM RSVA405 (AMPK agonist) for 24 h. In the si-Nek6 + tautomycetin group, cells were treated with 20 nM PP1 inhibitor tautomycetin for 1 h.
Cell apoptosis
Flow cytometry assay was performed to detect cell apoptosis. SH-SY5Y cells at logarithmic growth stage were inoculated with 1 × 104/well in a 6-well plate and resuspended with complete culture medium. 5 μL Annexin V FITC and 5 μL PI was added for 15 min according to the instructions of the Annexin V-FITC/PI double-staining kit (Beyotime). Analysis was performed by flow cytometry within 30 mim.
Cell activity
SH-SY5Y cells inoculated in 96-well plates were treated with OGD/R, 10 μL of MTT (5 g/L) solution was added and incubated for 4 h at 37 °C in a 5% CO2 incubator, and then 150 μL of DMSO was added and shaken for 10 min at room temperature. The absorbance value of each well at 490 nm was measured by using SynergyHT multifunctional microplate analyzer (Thermo Fisher Scientific).
Tandem fluorescently labeled LC3 (mRFP-GFP-LC3) detects autophagy flux
SH-SY5Y cells were cotransfected with a tandem expression vector mCherry-GFP-LC3 (Shanghai Genechem Co., Ltd.) and pcDNA-Nek6 vector. OGD/R treatment was performed 24 h after transfection and observed by fluorescence microscope. EGFP (enhanced green fluorescent protein) is acid-sensitive and will lose its fluorescence when autophagosomes fuse with lysosomes, which reduces the pH value; whereas, mCherry is acid-insensitive and positively sustained in both autophagosomes and autolysosomes.
Nek6 mRNA stability
Actinomycin D (ActD, Sigma) treatment was performed to detect the Nek6 mRNA stability. SH-SY5Y cells at logarithmic growth stage were treated with 5 μg/mL ActD, and total RNA of each group was extracted at 0, 1, 2, 3, and 4 h, respectively. Nek6 level was detected by qPCR.
Prediction of methylation sites in Nek6 sequences
Login SRAMP website (https://www.cuilab.cn/sramp), enter Nek6 transcript sequence (fasta format), obtain Nek6 sequence of N6-methyl adenosine (m6A) methylation modification site.
Methylated RNA immunoprecipitation (MeRIP)
Nek6 methylation level was detected by MeRIP assay by using a Magna RIP RNA-binding Protein Immunoprecipitation Kit (Millipore) in accordance with the manufacturer’s protocol. SH-SY5Y cells or tissues were lysed by RIP lysis buffer and incubated with antibodies against m6A (Synaptic Systems) at 4 °C overnight. IgG was used as negative control. The immunoprecipitated RNAs were eluted and analyzed by qPCR.
Co-immunoprecipitation (Co-IP)
After the cells were washed with PBS, 500 μL RIPA lysate was added for lysis on ice, and centrifuged at 12,000 r/min for 5 min. 50 μL supernatant of lysate was taken as input group, and the remaining 450 μL supernatant was added to Nek6 antibody as IP group, and incubated overnight at 4 °C. The next day, 20 μL protein A/G magnetic beads (Thermo Fisher Scientific) were added and incubated at 4 °C for 2 h. After washing five times with wash Buffer, 50 μL of 1 × loading buffer was added for denaturation at 95 °C for 5 min. Western blot was used to determine the protein interaction between PP1 and Nek6.
Statistical analyses
The cell experiment was repeated three times. SPSS 19.0 software was used for statistical analysis, and the measurement data was expressed as mean ± standard deviation (SD). Multi-group comparison was one way anova with tukey’s post-hoc, and two group comparison was unpaired two tailed t test. P < 0.05 was considered as statistically significant difference.




Comments (0)