
Remember me

Cardiovascular diseases (CVD) continue to be a widespread issue in the world of health, resulting in a substantial impact on sickness and death rates, as this ailment is responsible for about 33% of all fatalities [1]. CVD can be identified by a range of typical indicators such as discomfort in the chest, difficulty breathing, an irregular heart rate, exhaustion, and a decrease in overall physical endurance [2, 3]. CVD refers to a group of conditions that impact the functioning of the blood vessels and heart, such as atherosclerosis, diabetes, obesity, heart attacks, hyperhomocysteinemia, heart failure, high blood pressure (BP), and heart problems related to diabetes [4, 5]. The usual factors that are widely recognized as causing and aggravating cardiovascular disease, such as elevated levels of cholesterol and blood pressure, diabetes, and tobacco use, are still widely accepted as the main agents responsible for the onset and progression of this condition [6].
Persistent inflammation is a primary influence on the onset of heart disease, playing a vital role in its progression. Chronic inflammation can increase the progression of heart disease, making it a significant contributing factor to its development [7, 8]. The interplay between lipids and inflammation is a key defining feature of conditions related to atherosclerosis [9]. The initiation of inflammatory pathways, as well as their interaction with pathways that control the function of heart muscle, blood vessels, and connective tissue, are all part of the process that leads to heart failure [10].
Given the central role of inflammation in cardiovascular pathology, recent research has focused on the molecular sensors and pathways that initiate and sustain these responses—most notably, the inflammasome complexes.
Inflammasomes are collections of proteins present in cells that stimulate the synthesis and release of carefully regulated and highly inflammatory substances, including IL-18 and IL-1β. IL-1β is especially strong and can affect different types of cells, potentially initiating various inflammatory pathways by prompting the production of other cytokines like TNF-α, IL-8, and IL-6 in target cells [11]. IL-18 plays an important function that varies depending on the circumstances. In the presence of IL-18, IL-12enables the initiation of a type 1 immune response by prompting T and NK cells to produce INF-γ [12, 13]. If IL-12 is not present, IL-18 is capable of triggering type 2 immune reactions, which may include the creation of IL-4 and IL-13 as well as immunoglobulin E [14,15,16]. The inflammatory cytokines that rely on inflammasomes are potent substances with a diverse range of roles and are quickly and commonly triggered in both nonsterile and sterile inflammation . While immediate inflammasome activation is necessary for protecting the host against infections, it may also lead to detrimental consequences in cases of excessive or prolonged response .
The recent evidence has established inflammasomes—particularly the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome—as key regulators of sterile inflammation in cardiovascular disease [17, 18]. Inflammasomes act as intracellular sensors that detect metabolic danger signals such as oxidized LDL, cholesterol crystals, and mitochondrial dysfunction—factors commonly elevated in conditions like atherosclerosis, hypertension, and myocardial infarction [19, 20]. Upon activation, inflammasomes initiate a cascade that includes caspase-1 activation and the release of pro-inflammatory cytokines like interleukin (IL)-1β and IL-18, which contribute to endothelial dysfunction, cardiac remodeling, and plaque instability [21, 22]. This chronic inflammatory signaling not only promotes disease progression but also exacerbates tissue injury following ischemic events. As such, the inflammasome serves as a molecular link between immune activation and cardiovascular pathology, making it a promising target for therapeutic intervention.
Numerous research works have conclusively established the crucial function that exercise serves in controlling weight [23], blood sugar [24], BP reduction [25], and lipid regulation [26] consequently lowering the risk of CVD [27]. More specifically, structured exercise—a planned and repetitive form of exercise —has been shown to produce direct benefits on both cardiac structure and vascular function. Exercise enhances myocardial strength, improves cardiopulmonary capacity, promotes efficient blood flow, and increases cardiovascular resilience and overall heart health [6]. In addition, regular exercise, whether structured or incidental, exhibits anti-inflammatory effects [26]. Many people agree that exercise is an effective method for decreasing ongoing inflammation. This process involves inhibiting the creation of molecules that trigger inflammation while also encouraging the secretion of proteins that have anti-inflammatory properties [26]. Previous studies have indicated that engaging in consistent exercise can effectively diminish the activity of the NLRP3 inflammasome, resulting in a notable decrease in the manufacture of IL-18 and IL-1β, which are both cytokines associated with inflammation [27, 28].
Inflammation and cardiovascular diseasesAcute inflammation in cardiovascular eventsNumerous ailments are connected to the process of inflammation, encompassing conditions such as diabetes, obesity, and various types of infections. Moreover, inflammation is heightened in both short-term and long-term cardiac disorders, ranging from heart attacks to enlarged heart failure.
Acute myocardial infarction is a health condition characterized by the death of heart muscle cells caused by restricted blood flow to the heart, often caused by blockages in the coronary arteries. This condition often leads to elevated levels of GMCSF, TNFα, MCSF, GCSF, IFNβ, and IFNα in the plasma of affected patients [30]. Acute cardiac inflammation resulting from myocardial infarction is made possible through the contribution of macrophages, neutrophils, and mast cells, as these cells release pro-inflammatory cytokines [31]. After a heart attack, Neutrophils quickly gather at the injured area by releasing the protein IL-1β through the alarmin (S100A8/9)- Toll-like receptor (TLR) 4-inflammasome pathway. IL-1β attaches to receptor 1 on both hematopoietic stem cells and progenitor cells, causing an escalation in the production of granulocytes and a decline in cardiac function [32]. In addition to generating cytokines, mast cells also discharge histamine, which triggers inflammation in the heart's surrounding area after a myocardial infarction [31]. Furthermore, macrophages have a vital function in controlling inflammation after a myocardial infarction through the secretion of both anti- and pro-inflammatory cytokines [33,34,35]. Take, for instance, the macrophages that cause inflammation, which are most prevalent in the heart within the first 1–3 days after a MI. During this time, they release substances like GMCSF, TNF-α, IFN-γ, and IL-1β, which involve in the inflammatory response. In contrast, the anti-inflammatory macrophages take over 5–10 days after an MI and aid in tissue repair by releasing substances like TGF-β1, IL-10, and VEGF [33]. Macrophages, can be separated into different forms depending on the presence of specific proteins. These forms can be either pro-inflammatory or anti-inflammatory, influenced by the effects of certain molecules called cytokines. For example, cytokines such as IFNγ and GMCSF promote pro-inflammatory macrophage states, while others like IL-10, IL-4, TGFβ1, and IL-13 have the opposite effect and encourage anti-inflammatory polarization of macrophages [33]. Not only the nearby macrophages in the heart, but also the distant monocytes have a significant role in controlling inflammation following a cardiac event. Monocytes with increased levels of Ly6C are capable of turning into pro-inflammatory macrophages, and this transformation is aided by CCR2. Conversely, monocytes with lower Ly6C levels differentiate into anti-inflammatory macrophages through the involvement of CX3CR1, triggered by sudden cardiac stress [33]. Jung and et al. and Yang et al. uncovered the specific process by which IL-10 works against inflammation and dysfunction in the heart by converting macrophages into an anti-inflammatory state [34, 35]. To summarize, pro-inflammatory macrophages are the primary type found in the infarcted area after a MI. However, during the healing stage, anti-inflammatory macrophages become the dominant type in the infarcted region and the level of IL-10 also increases [35]. Similarly, the introduction of IL-10 following a mouse myocardial infarction led to improved recovery and performance of the heart due to the macrophages being transformed into anti-inflammatory cells [34]. In addition to the number of cytotoxic CD8 + T-cells and CD4 + helper T-cells infiltrating the tissue is higher in macrophages and neutrophils, and in hearts that are experiencing ischemia and hypertrophy. Research has proven that reducing these CD4 + T-cells using antibody therapy can lessen the severity of heart failure after a heart attack. On the other hand, introducing CD4 + cells from the spleen, along with CD3 + T-cells, using adoptive transfer techniques, researchers have discovered that fibrosis and hypertrophy can be induced in mice who do not have any pre-existing conditions [36]. During a heart attack, reduced oxygen levels and the restoration of blood flow can cause the immune cells, such as macrophages and neutrophils, to release inflammatory chemicals known as cytokines [37,38,39]. The role of inflammation in the progression of CVD in people with atherosclerosis has been suggested. However, by administering IL-1β antibody, the occurrence of MI and angina is significantly decreased, therefore providing evidence that targeting inflammation could effectively manage and prevent cardiovascular issues in these individuals [40]. The general invasion of immune cells that secrete cytokines after a heart attack is responsible for driving the alteration of the heart's structure. Therefore, the methods to decrease the number of infiltrating immune cells in the heart or decrease their activity in a failing heart could potentially be an effective treatment for ischemic heart damage. The enlargement of cardiomyocytes and the development of fibrosis are key features of cardiac hypertrophy and remodeling, ultimately leading to heart failure. Recent research has shown that inflammatory and immune cells pathways contribute with the advancement of nonischemic heart failure among patients [41]. The process of bringing in immune cells into the heart tissue leads to the development of enlarged heart muscles [42]. Preventing the recruitment of monocytes in the heart has a major effect on halting chronic remodeling caused by continuous stress. This has been proven by eliminating the MCP-1 gene or utilizing a MCP-1 neutralizing antibody, both of which successfully prevent the occurrence of chronic hypertrophic cardiac remodeling [43, 44].
Chronic inflammation and cardiac remodelingChronic inflammatory response is mediated by substances like IL-1β, TNFα, adiponectin, CRP, PAI-1, and IL-6, which contribute to lipid accumulation, atherosclerotic plaque development, and the progression of cardiovascular diseases such as coronary artery disease, myocardial infarction, and stroke [29].
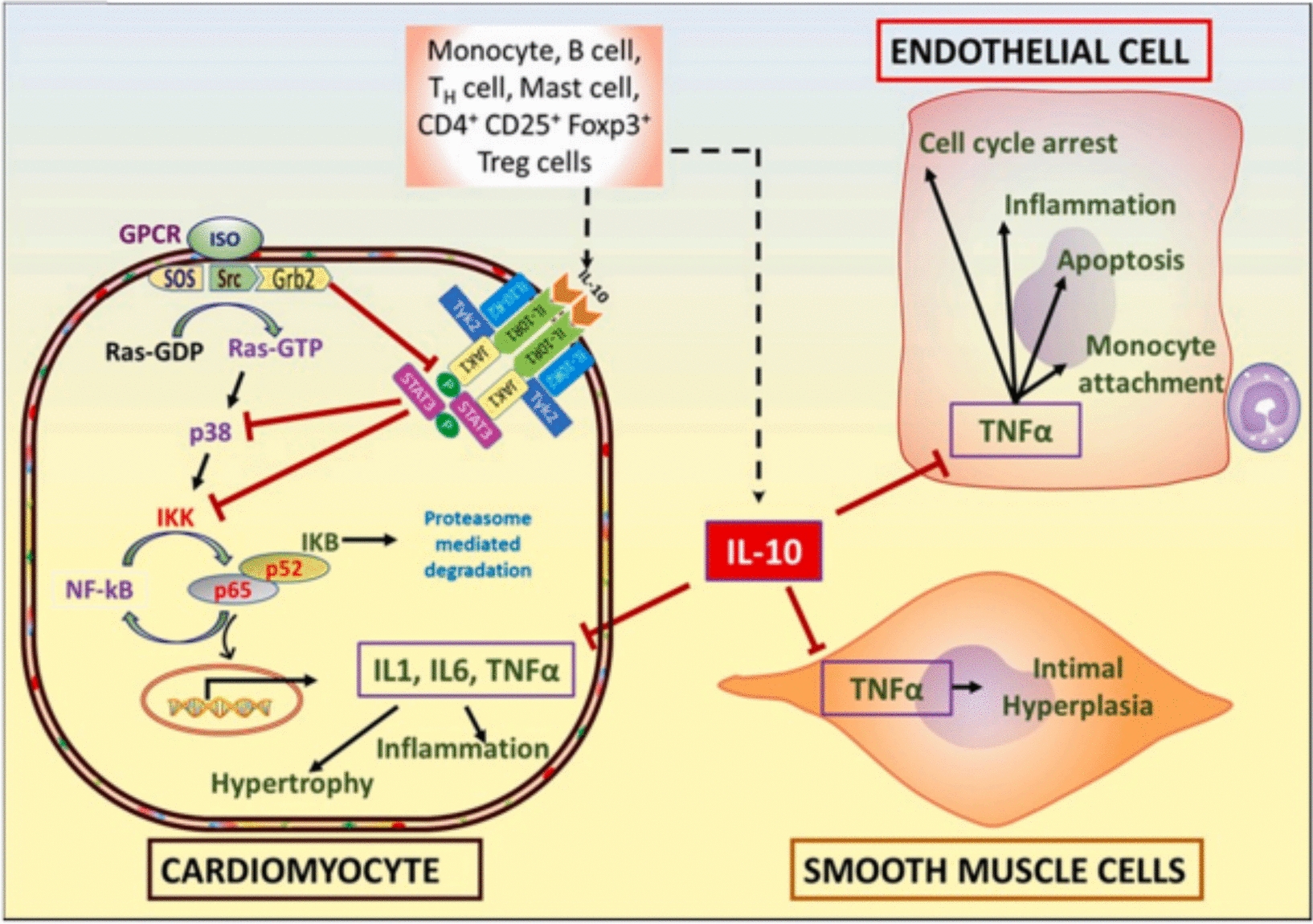
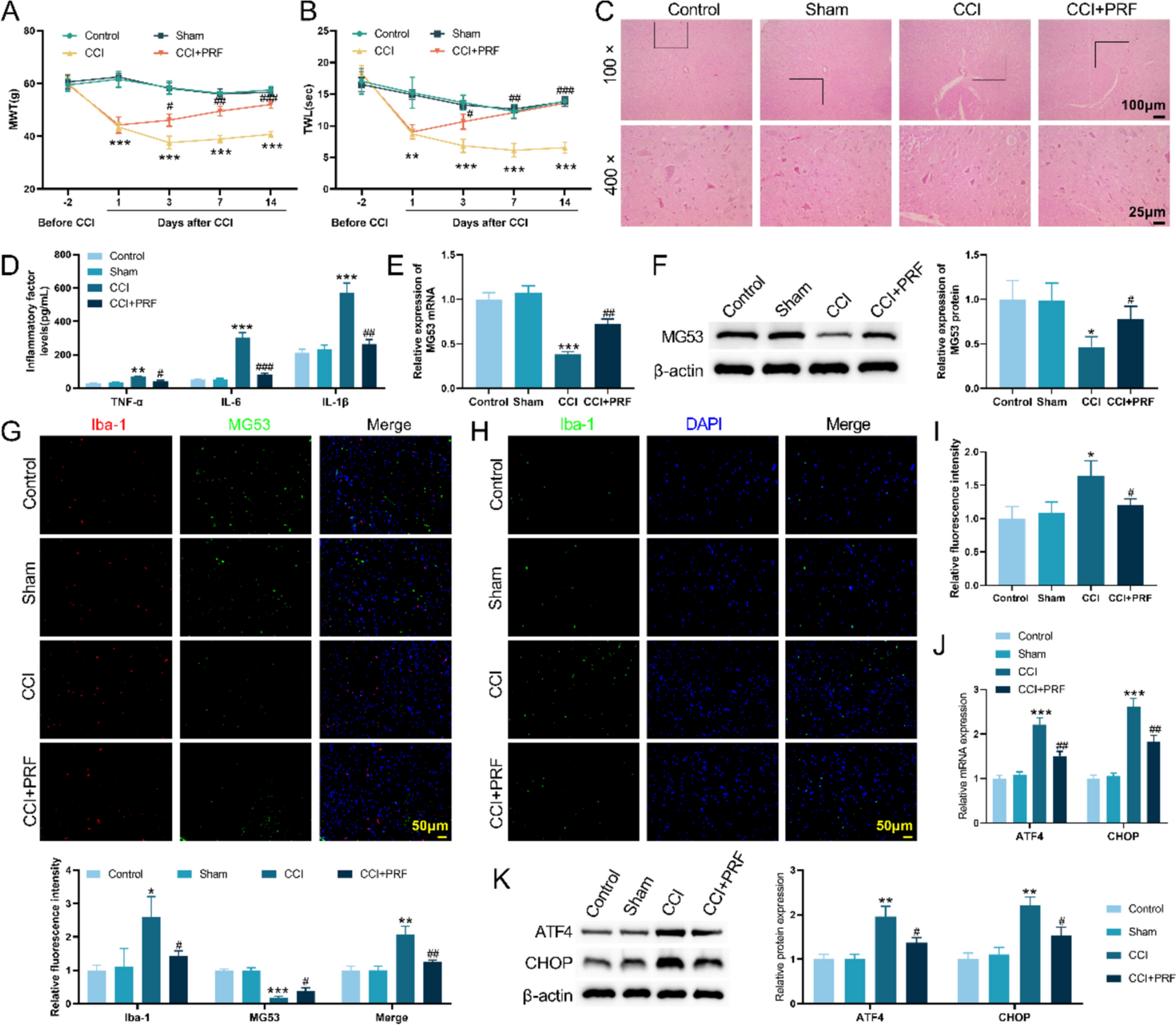
Chronic inflammation also contributes to progressive cardiac hypertrophy and fibrosis. T-cells producing IFNγ activate cardiac fibroblasts via integrin α4, leading to TGF-β-mediated fibrosis [41]. As anticipated, removing TCR-α from the genetic makeup results in the improvement of cardiac fibrosis and heart failure [41]. When the heart muscle is exposed to TAC, it increases ROS. This leads to a higher level of isolevuglandin (IsoLGs) that causes changes to proteins in the cardiac tissue. This process enables the activation of T-cell receptors, ultimately leading to the proliferation of T-cells. Curiously, the use of a cleansing agent for IsoLGs decreases T-cell proliferation and enhances heart function, suggesting that the overstimulation of T-cells via reactive oxygen species (ROS) may be a crucial contributor to the onset of cardiovascular problems following exposure to TAC [45]. Intriguingly, microbiomes possess the ability to manage cardiac remodeling using their metabolites, which is reliant on the existence of T-cells [46]. Individuals who have been diagnosed with dilated cardiomyopathy experience an increase in IL-2, IL-1α, TNFα, and IL-6 [30]. In the past, we have demonstrated that the expression of TNF-α and IL-1β was enhanced in cardiomyocytes treated with ISO. However, with the use of IL-10 treatment, there was a notable decline in the levels of these cytokines through the inhibition of NF-κB via STAT-3 [47] (Fig. 1). To sum up, the involvement of pro-inflammatory cytokines and inflammatory cells is necessary for the development of hypertrophic cardiac remodeling. As a result, utilizing IL-10, which is an anti-inflammatory treatment, could potentially improve cardiac function and lead to better outcomes for patients. Inflammaging, which is the persistent low-level inflammation seen in older individuals, is connected to issues such as impaired mitochondrial function, ongoing low-level endotoxin in the body, inflammation related to metabolism, imbalanced bacteria in the gut, and aging of immune cells and DNA. These factors ultimately contribute to age-related chronic illnesses like CVD [48,49,50]. As we age, various elements such as metabolites, modified proteins, micro RNAs, mitochondrial DNA, cytokines, and immune cells are discharged, resulting in the onset of inflammation within the body [48, 51,52,53]. Within the cardiac system, inflammaging has connections with numerous types of heart conditions, including atherosclerosis, elevated blood pressure, and hypertrophic remodeling of the heart muscle [54]. As one gets older, the immune cells of the body, including macrophages, neutrophils, B-cells, T-cells, and dendritic cells, work together to induce inflammation, which can have negative impacts on an individual's overall health [48,49,50, 55,56,57,58]. The main focus of research on heart inflammation has mainly centered on the involvement of monocytes/macrophages and T-cells. As people age, there is a decline in the population of protective anti-inflammatory macrophages (F4/80 + CD206 +), and an increase in the number of inflammatory macrophages originating from monocytes (F4/80 + CD206-) [59, 60]. In older mice, there is a notable rise in the amount of MCP-1, a protein that controls the influx of macrophages, in the left side of the heart. In addition, there is an increase in the presence of MMP-9, an enzyme produced by macrophages that contributes to the development of enlarged heart muscles and scarring [58]. MMP9 and MCP-1 serve as plasma biomarkers for the process of cardiac aging in mice. Further research should be conducted to examine their specific role in the aging of the human heart [58]. The presence of inflammatory indicators released by macrophages, such as TNFα, which are closely linked to cardiac inflammation, experiences a notable rise in the blood plasma of older mice [58]. As the mice grew older, there was a significant rise in the concentrations of MCP-2, MCP-1, and MIP-2 in their plasma. These biomarkers are directly linked to the left ventricular end-diastolic dimensions [58]. Removing the MMP9 gene results in a rise in anti-inflammatory macrophages within the hearts of older mice, resulting in improved conditions of myocardial fibrosis and diastolic dysfunction [60, 61]. An important discovery suggests that administering atorvastatin, a medication intended to lower lipid levels, result in a reduction in MMP9 levels and effectively inhibits the rise in left ventricle thickness, size of cardiomyocytes, accumulation of collagen, and the proportion of type I to type III collagen in aged rats [62]. Similarly, increasing the expression of MMP9 in macrophages results in an exaggerated growth of the heart, reduction in blood vessel density, heightened inflammation, and increased scarring in older mice [63]. Macrophage-produced MMP9 has a remarkable effect on the progression of age-related alterations in cardiac function. When MMP28, which promotes a macrophage profile that is anti-inflammatory, is removed through genetic means, the levels of MMP9 in the left ventricle of older individuals increase, resulting in a worsened state of inflammation and impairment in cardiac function [64]. As mice get older, their cardiac macrophages experience a decrease in the activity of inflammatory genes and a rise in the expression of fibrosis-related genes. This emphasizes the significant function of macrophages in the age-related inflammation of the heart [65]. The immune cells known as T-cells have been extensively researched in relation to cardiac inflammation. When CD4 + T cells from elderly individuals are stimulated in a laboratory setting, they exhibit characteristics of an inflammatory Th17 type, along with impaired mitochondrial function and deficiencies in autophagy. However, treatment with metformin can reverse these factors, bringing the Th17 phenotype back to a state resembling that of young people, while also improving mitochondrial function and addressing autophagy issues [66]. If the production of ROS is hindered, there is a decline in Th17 levels, indicating a potential link between mitochondrial and oxidative stress and Th17 activation in inflammation. Exploring the efficacy of metformin in mitigating cardiac inflammation and its effect on immune cells, such as monocytes and macrophages, in human physiology is advisable. Studies have revealed that older adults typically have shorter telomeres, which puts them at a higher risk for coronary heart disease. This implies a potential relationship between older age, aberrant white blood cell development, and cardiovascular disorders [67]. In older mice, the activation of dendritic cells in the lymph nodes and spleen has been found to trigger the maturation of Th1 and Th17 CD4 + T cells, resulting in the release of IFNγ and IL-17. This effect can be seen when aged dendritic cells are transplanted into young mice one week before a heart transplant, decreasing the chances of the allograft's survival. Notably, the use of senolytics (specifically, dasatinib and quercetin) inhibits the dendritic cells, ultimately decreasing the immunogenicity of Th1 and Th17 responses and resulting in extended survival of cardiac allografts [50]. As individuals and mice age, there is a greater presence of effector memory and terminally differentiated CD4 + T-cells in their bloodstream, while the amount of naive CD4 + T-cells decreases among older groups of both species [55]. When aged CD4 + T-cells are moved into NSG-DR1 mice lacking a sufficient immune system, it results in elevated levels of inflammation and cardiac fibrosis. This is accompanied by heightened activity in various inflammatory pathways such as cytokine–cytokine receptor interaction, IL-17 signaling, chemokine signaling, TNF signaling, NOD-like receptor signaling, and coagulation cascade and the complement [55]. When mice are exposed to human T-cells, it causes an escalation of macrophages, monocytes, and dendritic cells within their hearts. This indicates that T-cells have impact on orchestrating the infiltration of immune cells and causing heart inflammation [55]. Several experiments have shown that different varieties of immune cells, such as T-cells and dendritic cells, in the process of inflammaging in the heart. Consequently, utilizing an approach centered on immune cell manipulation may prove to be a viable method for controlling inflammaging in cardiac tissues.
Fig. 1
The anti-inflammatory cytokine IL-10 plays a crucial regulatory role in the functioning of cardiac cells. It is secreted by different lymph and immune cells and its main function is to counteract the effects of ISO-induced hypertrophy and TNFα-mediated signaling, both of which contribute to inflammation. Isoproterenol (ISO) triggers GPCR activation, leading to the production of Ras-GTP from Ras-GDP. This result in the activation of the p38 pathway, resulting in the increased expression of genes associated with hypertrophy and inflammation, through the stimulati
Comments (0)