
Remember me
Inspired by the problems of drug resistance, metastasis, and side effects of paclitaxel, oxaliplatin, and 5-FU and the advantages of the specificity and broad spectrum of natural drugs used in cancer treatment, we selected RKO cells, a poorly differentiated human colon cancer cell line sensitive to cell death, to screen for antitumor activity in a natural compound library. We incubated RKO cells with various compounds (10 μM) for 24 h and assessed their viability via a Cell Counting Kit-8 (CCK-8) assay. As shown in Fig. 1a, b, 13 compounds were identified as candidate compounds using the criterion of an antitumor activity exceeding 80%. Furthermore, we identified ACE as the best compound with broad-spectrum antitumor effects after measuring the semi-inhibitory concentrations (IC50) of the compounds in different tumor cell lines (Fig. 1c, d). Unexpectedly, we found that ACE is more effective against colorectal cancer cells, with IC50 values ranging from 1.4 μM to 1.9 μM, and is almost nontoxic to normal intestinal epithelial cells, suggesting that ACE is more suitable for colorectal cancer treatment (Fig. 1e–i).
Fig. 1
ACE inhibits cell migration and drug resistance. a Flow chart of the screening strategy. RKO cells were treated with a library of 420 natural compounds (10 μM) for 24 h. Cell viability was determined via a CCK-8 assay. The IC50 values of the candidates in different tumor cell lines were further examined. b Identification of antitumor compounds in the natural compound library for which the cell viability was lower than 20%. A change in color from blue to white represents a decrease in cell viability. c Structure of ACE. d IC50 values of ACE in different tumor cell lines. (n = 3, error bars represent SEM). e-h The inhibition ratio and IC50 of ACE (24 h) in HCT116 and RKO cells were measured via the CCK-8 assay. Inhibition ratios of NCM460 (f), SW620 (g), and HT29 (h) cells after ACE treatment for 24 h. (n = 3, error bars represent SEM). i Microscope images showing morphological changes in ACE-treated RKO and HCT116 cells. Scale bars, 200 μm. j, k RKO and HCT116 cells were incubated with ACE (0.1, 0.5, or 1 μM) in 12-well plates for 10 days, and the colony-forming potential of the tumor cells was assessed via crystal violet staining. (n = 3, error bars represent SEM, one-way ANOVA). l, m ACE (1, 2, or 5 μM) was incubated with RKO and HCT116 cells in 12-well plates for 24 h, and the proliferation of the tumor cells was detected via an EdU kit. (Scale bars, 200 μm. n = 3, error bars represent SEM, two-way ANOVA) n, o Cell cycle analysis and statistical analysis of RKO and HCT116 cells treated with ACE (1, 2, or 5 μM) for 24 h via flow cytometry. n = 3, data are shown as the mean ± SEM. The experiments consisted of three biological replicates with similar results. ****P < 0.0001
To determine the anti-proliferative effect of ACE, we conducted a colony formation assay and 5-ethynyl-20-deoxyuridine (EdU) assay and revealed that ACE significantly reduced colony formation efficiency and cell growth (Fig. 1j–m). In addition, flow cytometry revealed that ACE significantly induced G2/M phase arrest in HCT116 and RKO cells (Fig. 1n, o). Next, the Annexin V-FITC/PI double-staining assay revealed that ACE treatment induced cell death in HCT116 and RKO cells in a concentration-dependent manner, which was also confirmed by the calcein/PI double-staining assay (Supplementary Fig. 1a–d). In addition, ACE significantly inhibited the growth of oxaliplatin-resistant HCT116 cells (Supplementary Fig. 1e). Furthermore, ACE significantly decreased the migratory ability of CT26 and LoVo cells and downregulated the expression of EMT pathway proteins (Supplementary Fig. 1f–i). In conclusion, ACE inhibited cell proliferation, promoted cell cycle arrest, induced cell death, and inhibited cell migration and drug resistance.
ACE induces specific ferroptosis in colorectal cancer cellsAlthough ACE significantly inhibited cell proliferation and induced cell death, unlike previous reports, ACE did not activate the classical apoptotic pathway (Supplementary Fig. 1j, k). We further explored its potential non-apoptotic death mechanism. Next, we used a multiomics-based strategy (proteomics, transcriptomics, and metabolomics) to investigate the possible pathways of ACE-induced cell death. KEGG enrichment analysis of the proteomic data revealed that the ferroptosis pathway was the most enriched pathway, and the ubiquinone biosynthesis and unsaturated fatty acid biosynthesis pathways related to ferroptosis were also enriched in the ACE treatment group (Fig. 2a, b). In addition, GO enrichment analysis revealed that cellular components such as cell membranes and mitochondria were strongly associated with ferroptosis (Supplementary Fig. 2a, b). As expected, transcriptomic analysis revealed that the ferroptosis signaling pathway was significantly activated after ACE treatment (Fig. 2c and Supplementary Fig. 2c). On the basis of the important role of ferroptosis in overcoming tumor drug resistance, we tentatively hypothesized that ACE may induce ferroptosis in colorectal cancer cells.
Fig. 2
ACE induces ferroptosis in colorectal cancer cells. a, b Potential pathway analysis by KEGG enrichment in RKO (a) and HCT116 (b) cells and a heatmap showing differentially expressed proteins. Ferroptosis-related genes were labeled (fold change ≥1.2, unique peptides ≥2). c Volcano plot showing ferroptosis pathway gene expression in ACE-treated MCF7 cells (fold change ≥2, p value ≤ 0.05). d Heatmap showing oxidized polyunsaturated fatty acids after ACE (5 μM) treatment for 12 h. e AA and their metabolite levels in RKO cells treated with ACE (5 μM) for 12 h compared with DMSO. (n = 3, error bars represent SEM, Student’s t-test). f-i Viability of RKO and HCT116 cells treated with ferroptosis inhibitors (Fer-1: ferrostatin-1; Lip-1: liproxstatin-1; DFO: deferoxamine mesylate). (f) Z-VAD-FMK (g), NEC-1 (h), and CQ (i) after treatment with ACE (2 μM). The data are shown as the mean ± SEM. (n = 3, error bars represent SEM, one-way ANOVA). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns not significant
Since lipid peroxidation is essential for ferroptosis, we next analyzed the changes in the levels of oxidized fatty acids and redox agents in RKO cells in the presence or absence of ACE. First, we observed that ACE treatment led to a significant increase in the levels of oxidized polyunsaturated fatty acids, particularly arachidonic acid (AA) and linoleic acid (LA), reflecting the accumulation of intracellular lipid peroxidation products (Fig. 2d, e and Supplementary Fig. 2d–f). Second, antioxidants were significantly elevated, suggesting that ACE-mediated lipid peroxidation may rely on increased synthesis rather than decreased scavenging (Supplementary Fig. 2g). Notably, the levels of nonenzymatic metabolites such as 9-HODE and 12-HDHA increased significantly after ACE treatment (Supplementary Fig. 2h). The multiomics results also revealed that ACE significantly regulated HO-1 and glutamate-cysteine ligase modifier subunit (GCLM) (Supplementary Fig. 2i). Furthermore, as shown in Fig. 2f–i, the cytotoxicity of ACE was blocked by ferroptosis inhibitors but not by apoptosis, necroptosis, or autophagy inhibitors. These results suggest that ACE-induced specific ferroptosis is a key determinant of colorectal cancer cell death and is largely dependent on Fe2+.
ACE induces canonical and noncanonical ferroptosisOn the basis of the significant enrichment of the ferroptosis pathway, we verified whether ACE induced ferroptosis by examining morphological and biochemical characteristics. In contrast to other forms of cell death, lipid peroxidation is a crucial marker of ferroptosis. To investigate the level of ACE-mediated lipid peroxidation, two fluorescent probes, BODIPY-C11 and Liperfluo, were used in this assay. BODIPY-C11 staining revealed that ACE promoted lipid peroxidation more strongly than the ferroptosis inducers RSL3 and erastin did, and this effect was reversed by the iron chelator DFO and the lipid peroxidation scavenger Lip-1 (Fig. 3a, b, and Supplementary Fig. 3a–c). Moreover, ACE significantly increased the level of intracellular lipid peroxidation in a concentration-dependent manner (Fig. 3c and Supplementary Fig. 3d–i). In addition, ACE generally elevated lipid peroxidation levels in other colorectal cancer cells, including drug-resistant colorectal cancer cells (Supplementary Figs. 3j–l, and 4a–d). In addition, we detected a significant increase in the intracellular level of MDA, a cytotoxic product produced by lipid peroxidation metabolism (Fig. 3d and Supplementary Fig. 5a). Moreover, transmission electron microscopy revealed smaller mitochondria, increased membrane density, and fewer cristae, which indicate the morphological signature of ferroptotic cells (Fig. 3e). Additionally, ACE dose-dependently disrupted mitochondrial function, concomitant with significant inhibition of basal and maximal cellular respiration (Fig. 3f and Supplementary Fig. 5b, c). These results indicate that ACE treatment significantly increases the accumulation of lipid peroxides, subsequently inducing ferroptosis in colorectal cancer. Taken together, these data demonstrate that ACE induces canonical ferroptosis.
Fig. 3
ACE induces ferroptosis by inactivating GPX4 and accumulating Fe2+. a, b BODIPY-C11 staining (a) and flow cytometry (b) showing RKO cells treated with ACE (5 μM) with or without a ferroptosis inhibitor (Lip-1, DFO) or ferroptosis inducer (RSL3, erastin) for 2 h. (n = 3, scale bars, 1000 μm). c Liperfluo staining for analysis of lipid peroxidation in RKO and HCT116 cells after treatment with ACE (1, 2, or 5 μM) for 2 h. (n = 3, scale bars, 1000 μm). d Malondialdehyde (MDA) assay showing the lipid peroxidation levels of RKO cells treated with ACE. (n = 3, error bars represent SEM, one-way ANOVA). e Transmission electron microscopy images of RKO cells treated with ACE (5 μM, 12 h) and DMSO (12 h). 4200×: for the observation of intact individual cell morphology. 10500×: for the observation of clear mitochondrial morphology. Red arrowheads, mitochondrial atrophy with reduced cristae; black arrowheads, normal mitochondria. Seven cells per treatment condition were examined; scale bars, 5 μm. f Mitochondrial respiration was measured in ACE-treated (0.5 and 2 μM, 12 h) and DMSO-treated (Ctrl) RKO cells via a Seahorse XF96 system. The cells were treated with the indicated reagents (oligo: oligomycin, ATP synthase inhibitor). FCCP, mitochondrial oxidative phosphorylation uncoupler. Rot/AA: Rotenone and antimycin A, a mitochondrial respiratory chain inhibitor, were used to measure the basal oxygen consumption rate (OCR) and maximal respiration. (n = 3, error bars represent SEM, two-way ANOVA). g FerroOrange staining showing RKO cells treated with ACE (5 μM) with or without a ferroptosis inhibitor (Lip-1, DFO) or ferroptosis inducer (RSL3, erastin) for 24 h (n = 3, scale bars, 1000 μm). h Quantitative analysis of Fe2+ levels in RKO cells treated with ACE via a ferrous ion colorimetric assay. (n = 3, error bars represent SEM, one-way ANOVA). i HO-1 protein levels in RKO and HCT116 cells after dose-dependent ACE treatment. j DCFH-DA staining showing the intracellular ROS levels in RKO and HCT116 cells treated with ACE (1, 2, or 5 μM) for 2 h. (n = 3, scale bars, 200 μm). k RKO cells were treated with ACE (2, 5 μM) with or without the ROS scavenger NAC for 24 h, and cell viability was assayed via a CCK-8 assay. (n = 3, error bars represent SEM, one-way ANOVA). **P < 0.01, ***P < 0.001, ****P < 0.0001; ns not significant
To detect free intracellular ferrous ions (Fe2+), the fluorescent probe (FerroOrange) and Ferrous Ion Content Assay Kit were used. As shown in Fig. 3g, h and Supplementary Fig. 5d–k, ACE treatment resulted in a significant dose-dependent increase in Fe2+ in RKO and HCT116 cells, and DFO, but not Lip-1, significantly reversed the effect of ACE on increasing Fe2+ levels. Surprisingly, compared with the ferroptosis inducers RSL3 and erastin, ACE significantly increased the intracellular Fe2+ levels (Fig. 3g and Supplementary Fig. 5e). Moreover, the multiomics results revealed that the most significantly upregulated gene was HO-1, which is associated with iron metabolism (Supplementary Fig. 2i). This finding was subsequently confirmed by Western blot experiments (Fig. 3i and Supplementary Fig. 6a). When free intracellular ferrous ions accumulate in the cytoplasm, ROS are generated via the Fenton reaction, subsequently leading to ferroptosis in tumor cells. As shown in Fig. 3j and Supplementary Fig. 6b–d, ROS levels increased significantly after ACE treatment and caused cell death, whereas the ROS inhibitors N-acetyl-L-cysteine (NAC) and GSH reversed ACE-induced cell death (Fig. 3k and Supplementary Fig. 6e). These results suggest the important role of Fe2+ in ferroptosis induced by ACE (noncanonical ferroptosis). Taken together, these findings indicate that ACE induces ferroptosis via both canonical and noncanonical pathways.
ACE suppresses colorectal tumor growth in vivoAlthough ACE significantly inhibited GPX4 and elevated Fe2+ in colorectal cancer cells, thereby inducing ferroptosis, the tumor cytotoxicity of this compound in vivo is unknown. To verify the potential in vivo antitumor activity of ACE, mice subcutaneously inoculated with HCT116-luc tumors were treated with either corn oil or ACE orally once daily for 22 days (Supplementary Fig. 7a). The tumor fluorescence intensities in the 25 and 50 mg/kg groups were 57.5% and 36.95% of the control group, respectively (Fig. 4a and Supplementary Fig. 7b). In addition, treatment with ACE at 25 and 50 mg/kg significantly reduced tumor size, volume, and weight compared to the control group (Fig. 4b–f). There was no significant difference in weight among the different groups, and the immunohistochemical results also revealed no obvious toxicity to the heart, liver, spleen, lung, or kidney (Fig. 4d and Supplementary Fig. 7c). These results showed that ACE treatment significantly inhibited HCT116 tumor growth in mice. We subsequently seeded RKO cells into nude mice and administered ACE when the tumor volume approached 50 mm3. Similar to its efficacy at the cellular level, ACE was more effective in RKO tumor-bearing mice than in HCT116 mice (Fig. 4g–k).
Fig. 4
ACE induces ferroptosis in vivo. a Bioluminescence images of HCT116-luc tumors taken every week. b, g Xenograft tumor images of HCT116-luc (b) and RKO (g) fully grown tumors versus residual tumors treated with ACE (10, 25, and 50 mg/kg). c, h Tumor weights of (b) and (g). (n = 5, error bars represent SEM, one-way ANOVA). d, i Mouse body weight. (n = 5, error bars represent SEM, two-way ANOVA). e, j Tumor volume curves showing mice treated orally with ACE (10, 25, or 50 mg/kg) and measured every 2 days. (n = 5, error bars represent SEM, two-way ANOVA). f, k Tumor volume curves for each mouse in different treatment groups corresponding to (e) and (j), respectively. l Immunohistochemistry (IHC) images of Ki-67, cleaved caspase 3 (Cl-Cas-3), PCBP1, PCBP2, and GPX4 in corn oil- or ACE-treated (10, 25, or 50 mg/kg) HCT116-luc mice. Scale bar, 200 μm. m Quantification of tumor MDA levels in RKO mice after ACE (10, 25, or 50 mg/kg) treatment for 22 days. (n = 3, error bars represent SEM, one-way ANOVA). n Quantification of tumor Fe2+ levels in the corn oil or ACE (50 mg/kg) groups. (n = 3, error bars represent SEM, two-tailed unpaired Student’s t test). **P < 0.01, ***P < 0.001, ****P < 0.0001; ns not significant
Immunoblotting analysis of tumor tissues revealed that the number of Ki67-positive cells in ACE-treated tumors was significantly lower than that in the control group. Furthermore, ACE did not activate caspase-3, suggesting that ACE suppressed mouse tumor growth independent of apoptosis (Fig. 4l and Supplementary Fig. 7d). To determine whether ACE induced ferroptosis in tumor tissues, we measured the levels of the lipid peroxidation products MDA and Fe2+ and found that ACE elevated the levels of MDA and increased the accumulation of Fe2+ in mouse tumor tissues (Fig. 4m, n and Supplementary Fig. 7e, f). These results suggest that ACE induces ferroptosis in tumor cells and effectively inhibits colorectal cancer.
ACE elevates Fe2+ levels independent of HO-1Previous studies have suggested that elevated HO-1 may be a source of intracellular Fe2+.32 To elucidate the mechanism by which ACE elevates Fe2+, we further determined whether ACE increased intracellular Fe2+ by upregulating HO-1. Unexpectedly, ACE treatment significantly increased the intracellular Fe2+ levels within 12 min, but western blot analysis revealed that ACE did not upregulate HO-1 expression within 1 h (Supplementary Fig. 8a–d). Moreover, we cotreated cells with ACE and Znpp, an inhibitor of HO-1. However, no significant difference in cell viability was found, suggesting that ACE did not induce ferroptosis by activating the enzymatic activity of HO-1 (Supplementary Fig. 8e, f). Furthermore, ZnPP did not reverse the increase in Fe2+ levels induced by ACE (Supplementary Fig. 8g, h). This conclusion was further confirmed by using hemin, an agonist of HO-1 enzymatic activity (Supplementary Fig. 8g, i, j). Moreover, the changes in the ROS levels were not reversed by ZnPP or hemin (Supplementary Fig. 9a–d). These results indicate that ACE increases intracellular Fe2+ levels independently of HO-1. In addition, we found that cells appeared green after ACE treatment, suggesting that elevated intracellular Fe2+ plays an important role in ferroptosis induced by ACE (Supplementary Fig. 9e).
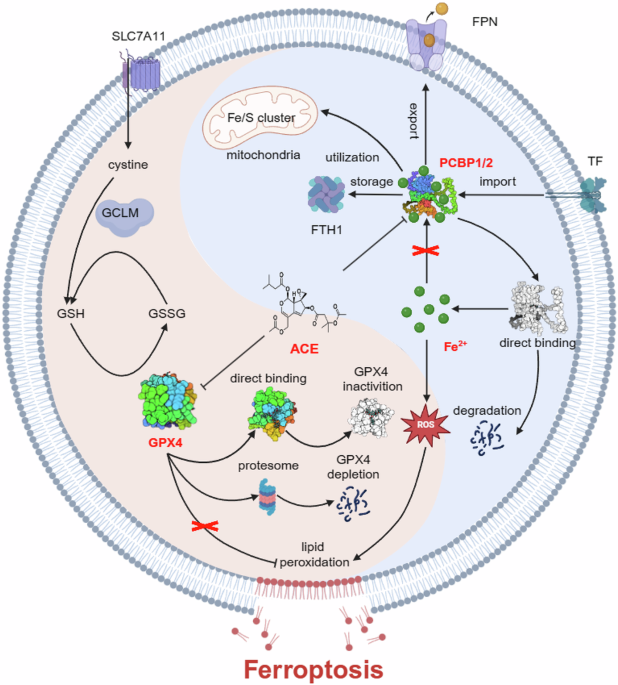
Downregulation of PCBP1/2 mediates Fe2+ release and induces ferroptosisTo explore the potential mechanism by which ACE elevates Fe2+ levels, iron metabolism proteins associated with ACE were screened by Western blot based on proteomic results (Fig. 5a, b and Supplementary Fig. 10a–c). Furthermore, we used drug affinity responsive target stability (DARTS), a technique for detecting the binding of small-molecule drugs to their target proteins, to explore potential target proteins of ACE. DARTS predicted 240 potential target proteins (intensity ratio ≥ 1.2), and four genes, namely, TF, GCLM, PCBP1, and SLC39A14, were directly associated with ferroptosis (Fig. 5c). Notably, PCBP1 or PCBP2, which have similar functions in the regulation of iron metabolism, were enriched in two assays. Consistent with the multiomics results, PCBP1 and PCBP2 proteins were significantly downregulated in a dose-dependent and time-dependent (within 1 h) manner (Fig. 5a, b and Supplementary Fig. 10d–i). Therefore, we speculate that ACE may increase the release of Fe2+ through PCBP1 and PCBP2.
Fig. 5
PCBP1/2 mediates ACE-induced Fe2+ accumulation and ferroptosis. a, b Western blot analysis of PCBP1 and PCBP2 protein levels in RKO and HCT116 cells after treatment with dose-dependent (a) or time-dependent (b) ACE. c Heatmap showing DARTS in the total protein of RKO cells treated with ACE for 1 h. Ferroptosis-related genes were labeled (fold change ≥1.2, unique peptides ≥2). d PCBP1/2 and Fe2+ levels in RKO cells were detected after ACE treatment for 24 h. The nuclei were stained with DAPI (blue), PCBP1/2 (green) was stained with a fluorescence-conjugated PCBP1/2 antibody, and Fe2+ was stained with FerroOrange (red) (n = 3 wells of a 12-well plate from one representative experiment; scale bars, 200 μm). e, f ROS levels (e) and Fe2+ levels (f) in PCBP1- or PCBP2-knockdown RKO and HCT116 cells were analyzed via flow cytometry. g Quantification of (f). (n = 3, error bars represent SEM, one-way ANOVA). h, i Flow cytometry analysis of Fe2+ levels in ACE-treated (1, 2, 5 μM, 24 h) PCBP1- or PCBP2-knockdown RKO cells, and the Fe2+ levels were quantified in (i). (n = 3, error bars represent SEM, one-way ANOVA). j Cell death analysis of PCBP1- or PCBP2-knockdown RKO and HCT116 cells via flow cytometry. k Western blot showing the overexpression efficiency of PCBP1 and PCBP2 in RKO cells, and the quantitative results are shown. (n = 3, error bars represent SEM, unpaired two-tailed Student’s t test). l, m Flow cytometry analysis of Fe2+ levels (l) and lipid ROS levels (m) in ACE-treated PCBP1- or PCBP2-overexpressing RKO cells. n Cell proliferation analysis of OE NC, OE PCBP1, and OE PCBP2 RKO cells (n = 3, SEM, two-way ANOVA). o Cell viability analysis showing the dose-dependent toxicity of ACE (0.5, 1, 2, 5, and 10 μM) in OE NC, OE PCBP1, and OE PCBP2 RKO cells via a CCK-8 assay. (n = 3, SEM, two-way ANOVA). *P < 0.05, **P<0.01, ***P<0.001, ****P < 0.0001; ns not significant
To verify the relationship between the elevated Fe2+ level of ACE and PCBP1/2, we performed immunofluorescence staining and found that PCBP1 and PCBP2 were significantly negatively correlated with intracellular Fe2+ (Fig. 5d and Supplementary Fig. 11a). We next designed a specific small interfering RNA (siRNA) to mimic the pharmacological inhibition of PCBP1/2 by ACE (Supplementary Fig. 11b, c). Interestingly, knockdown of PCBP1 and PCBP2 significantly elevated Fe2+ and ROS levels (Fig. 5e–g and Supplementary Fig. 11d, e). Furthermore, ACE did not significantly increase Fe2+ levels in cells lacking PCBP1/2 (Fig. 5h, i and Supplementary Fig. 11f–h). Moreover, we observed extremely poor cell status after knockdown, and the flow cytometry results indicated increased cell death in colorectal cancer cells (Fig. 5j and Supplementary Fig. 12a). These results suggest that ACE may induce ferroptosis by increasing Fe2+ and ROS via PCBP1 and PCBP2. As expected, the overexpression of PCBP1 and PCBP2 significantly inhibited the ability of ACE to increase lipid peroxidation and Fe2+, which further confirmed our speculation (Fig. 5k–m and Supplementary Fig. 12b–e). Moreover, cell proliferation was significantly promoted, and the cytotoxicity of ACE was significantly inhibited when PCBP1 and PCBP2 were overexpressed in RKO cells (Fig. 5n, o). These results suggest that ACE induces ferroptosis in colorectal cancer through PCBP1 and PCBP2.
Binding of ACE to PCBP1/2 induces ferroptosisTo investigate whether ACE directly binds to PCBP1 and PCBP2, we used a cellular thermal shift assay (CETSA) to detect whether the thermal stability of the proteins changed after ACE treatment. As shown in Fig. 6a, b, the PCBP1 and PCBP2 proteins were less thermally stable after ACE treatment, which was consistent with the results of DARTS. Furthermore, we determined the affinity between PCBP1 and ACE via surface plasmon resonance (SPR), and the results revealed that ACE bound directly to PCBP1 with a KD value of 0.8464 μM (Fig. 6c). Additionally, ACE dissociates slowly from the PCBP1 protein, suggesting that ACE binds to the PCBP protein in a more stable manner (Fig. 6c). In addition, DTT, a cysteine-rich thiol donor, could compete for cysteine-dependent ACE binding to PCBP1/2. As shown in Fig. 6d, e, the downregulation of PCBP1/2 and the increase in cytotoxicity caused by ACE were partially reversed by the addition of excess DTT, indicating that ACE binds directly to cysteine residues of PCBP. We then used Molecular Operating Environment (MOE) software to model the covalent binding of ACE with PCBP1/2 via its cysteine residues. Docking simulations were performed to explore the binding mode of ACE to each cysteine residue site of PCBP1 (AlphaFold ID AF-Q15365-F1) and PCBP2 (AlphaFold ID AF-Q15366-F1). As shown in Fig. 6f, g stable hydrogen bonds were formed between Cys54 and the backbone of ACE, suggesting that Cys54 is a possible binding site for both PCBP1 and PCBP2. In addition, Cys293 is another possible binding site for PCBP1. To further confirm the direct binding of ACE to PCBP1/2, we overexpressed wild-type PCBP1/2, the Cys54 mutant PCBP1, the Cys293 mutant PCBP1, and the Cys54 mutant PCBP2 with a GFP tag and subsequently analyzed direct binding via microtiter thermophoresis (MST). As expected, ACE bound strongly to PCBP1/2, with estimated Kd values of 4.64 nM and 204 nM. When Cys293 was mutated to alanine, its Kd became 325 times than that of wild-type PCBP1, whereas when Cys54 was mutated to alanine, ACE binding could not be detected in either PCBP1 or PCBP2 (Fig. 6h, i). Consistently, we found that the cytotoxicity of ACE was reduced in RKO cells overexpressing PCBP1/2 C54A (Fig. 6j). These results suggest that ACE elevates Fe2+ levels to induce ferroptosis in colorectal cancer through direct binding to PCBP1 and PCBP2.
Fig. 6
ACE directly binds to PCBP1/2. a, b CETSA showing the effect of ACE on the thermal stability of the PCBP1/2 protein in RKO cells. (n = 3, error bars represent SEM, two-way ANOVA). c Surface plasmon resonance (SPR) assay to determine the affinity between PCBP1 and ACE. d, e RKO cells were treated with ACE (2 or 5 μM) with or without the cysteine-rich thiol donor DTT for 24 h, after which cell viability (d) and protein levels (e) were detected. (n = 3, error bars represent SEM, one-way ANOVA). f, g Molecular modeling simulation of ACE docked on AlphaFold-predicted PCBP1 (f) (AF-Q15365-F1) and PCBP2 (g) (AF-Q15366-F1). h, i MST assay showing the binding of predicted binding site mutants, wild-type PCBP1 (h) or PCBP2 (i) to ACE. j Cell viability analysis showing the dose-dependent toxicity of ACE (0.5, 1, 2, 5, and 10 μM) in OE NC, OE PCBP1-C54A, and OE PCBP2-C54A RKO cells via a CCK-8 assay. (n = 3, error bars represent SEM, two-way ANOVA). ****P < 0.0001; ns not significant
ACE acts as a natural class II ferroptosis-inducing agentAs the upregulation of a series of antioxidant genes and factors was observed in the multiomics data, we examined the changes in the expression of several antioxidant genes. Consistent with the multiomics results, ACE significantly upregulated the expression of nuclear factor erythroid derived 2-like 2 (Nrf2) and its downstream target genes (e.g., SLC3A2, SLC7A11, and GCLM), ferroptosis suppressor protein 1 (FSP1), and dihydroorotate dehydrogenase (DHODH) (Supplementary Fig. 13a, b). Moreover, we found that the expression of GPX4, a core protein of the ferroptosis defense mechanism, was significantly downregulated (Fig. 7a, b and Supplementary Fig. 13c, d). Inactivation of GPX4 can occur through two processes: depletion of intracellular glutathione (GSH) or direct targeting of GPX4. As shown in Fig. 7c and Supplementary Fig. 2h and13e, ACE treatment increased the levels of GSH and cystine in RKO cells. Furthermore, ACE rapidly increased ROS levels and inhibited GPX enzyme activity, indicating that ACE, like RSL3, may bind directly to GPX4 (Fig. 7d and Supplementary Fig. 13f–h). As anticipated, the lack of GPX4 significantly promoted lipid peroxidation accumulation and cell death (Fig. 7e, f and Supplementary Fig. 13i–m). However, GPX4 overexpression increased the IC50 value of ACE in RKO cells (Fig. 7g and Supplementary Fig. 13n, o). Excess DTT reversed the downregulation of GPX4, confirming the binding between ACE and the cysteine residues of GPX4 (Fig. 7h and Supplementary Fig. 14a, b). As shown in Fig. 7i, we similarly performed molecular docking simulations and found that the active site, selenocysteine (U46), was the most favorable site for the covalent binding of GPX4 by ACE (Protein Data Bank ID 6NH3). Additionally, the thermal stability of the GPX4 protein increased upon treatment with ACE and RSL3 (positive control), indicating that ACE can bind to and stabilize GPX4 (Fig. 7j and Supplementary Fig. 14c). Finally, we performed MST assays of GFP-tagged GPX4 in the wild-type and disruptive mutant U46. The Kd value for ACE binding to the GPX4 protein was estimated to be 196 nM (Fig. 7i). However, once U46 was mutated to alanine, the binding between GPX4 and ACE was lost, which further diminished the cytotoxicity of ACE (Fig. 7i and Supplementary Fig. 14d, e). These results suggest that ACE inactivates GPX4 through direct binding to U46 in GPX4, triggering lipid peroxidation and subsequent ferroptosis.
Fig. 7
ACE induces GPX4 depletion for ferroptosis. a, b Western blot analysis of GPX4 expression in RKO and HCT116 cells after treatment with dose-dependent (a) or time-dependent (b) ACE. c, d Measurement of GSH levels (c) and GPX enzyme activity (d) in RKO cells after treatment with ACE (1, 2, or 5 μM) for 24 h. (n = 3, error bars represent SEM, one-way ANOVA). e, f Flow cytometry analysis of cell death (e) and lipid peroxidation (f) in GPX4-knockdown or WT RKO cells. g Dose-dependent toxicity of ACE in RKO cells overexpressing GPX4, and CCK-8 assays were used to measure cell viability. (n = 3, error bars represent SEM). h Western blot analysis of RKO cells treated with ACE (2 or 5 μM) with or without DTT for 24 h. i Molecular docking of ACE to GPX4 (6nh3). GFP-tagged wild-type or U46 disruptive mutant GPX4 proteins were maintained at constant concentrations and fluorescence intensities, and ACE was diluted in a 1/2-fold gradient. The MST-on time of 1.5 s and dissociation constants Kd were determined. j Thermal stability analysis of GPX4 protein interactions with the indicated compounds from 43 °C to 61 °C. k, l CHX analysis of GPX4 abundance in RKO cells treated with or without ACE (5 μM) for different durations was performed by immunoblotting, and the GPX4 intensity was quantified in (l). (n = 3, error bars represent SEM, two-way ANOVA). m Immunoblotting was used to detect the degradation of GPX4 in RKO cells induced with lysosome (Baf) and proteasome (MG132) inhibitors. n IP assay showing the ubiquitin (Ub) modification of GPX4 in RKO cells treated with ACE (5 μM) for 1 h, after which MG132 was added and incubated for 3 h. o CCK-8 assay showing the dose-dependent toxicity of ACE in RKO cells transfected with si-NC or si-GPX4. p Flow cytometry analysis of Fe2+ levels in GPX4-knockdown RKO cells. *P < 0.05, ****P < 0.0001; ns not significant
Next, we investigated the mechanisms by which ACE triggered GPX4 protein degradation. Western blot and real-time PCR results revealed that ACE downregulated not only GPX4 expression but also its mRNA levels (Fig. 7a, b and Supplementary Figs. 13c, S14f). Here, we explored mainly the posttranslational regulation of GPX4 since its protein levels decrease more rapidly than its RNA levels (Fig. 7b and Supplementary Figs. 13d, 14g). As shown in Fig. 7k, l, in combination with the protein translation inhibitor cycloheximide (CHX), the half-life of GPX4 in ACE-treated cells was faster than that in untreated cells, suggesting that ACE degraded GPX4 mainly at the protein level. In addition, Western blot experiments demonstrated that the proteasome inhibitor MG132 reversed the degradation of GPX4 by ACE but not by CQ (Fig. 7m and Supplementary Fig. 14h). The immunofluorescence results also revealed that the number of lysosomes did not increase (Supplementary Fig. 14i). Moreover, IP assays revealed that ACE increases the ubiquitination of GPX4 (Fig. 7n). These results suggest that ACE mediates the degradation of GPX4 through the ubiquitin-proteasome pathway and induces ferroptosis in colorectal cancer cells.
Finally, the cytotoxicity of RSL3 disappeared, whereas the cytotoxicity of ACE was partially reduced when GPX4 was knocked down since RSL3 did not increase Fe2+ (Fig. 7o, p and Supplementary Fig. 14j, k). These findings explain why ACE was more cytotoxic than RSL3 in oxaliplatin-resistant HCT116 cells (Supplementary Fig. 1e). Furthermore, PCBP1, PCBP2, and GPX4 synergistically promoted ferroptosis in colorectal cancer cells (Supplementary Fig. 14l, m). As shown in Fig. 4l, GPX4, PCBP1, and PCBP2 protein levels were reduced in tumor tissues in a dose-dependent manner, which was consistent with the results of the in vitro experiments. These results emphasize the effectiveness of the dual mechanism of ACE in inducing ferroptosis in colorectal cancer cells, which inactivates GPX4 and downregulates PCBP1/2 to release Fe2+.
Protein expression levels of PCBP1/2 and GPX4 in human tumor tissueTo validate the potential of the above models for clinical tumor treatment, we compared the tumor inhibitory effects of ACE with those of clinical first-line drugs and ferroptosis-positive drugs in xenograft models. We found that the tumor inhibitory effect of ACE was significantly greater than that of the ferroptosis-positive drugs sorafenib and artemisinin and even better than that of the clinical first-line drugs capecitabine and TAS-102, since ACE elevated Fe2+ and reduced PCBP2 and GPX4 significantly better than the other treatments did (Fig. 8a–d, Supplementary Fig. 15a–d). Although ACE induces ferroptosis in tumor cells by targeting PCBP1/2 and GPX4, previous studies have shown that ferroptosis may cause acute liver injury and acute kidney injury. To evaluate the safety of ACE for tumor therapy, we further conducted acute toxicology experiments in which mice were orally administered ACE (50 mg/kg) daily for 10 days. As shown in Supplementary Table 1, there were no significant changes in the blood parameters of mice, such as red and white blood cell counts or platelet counts; indicators of liver and kidney injury, such as ALT, AST, BUN, Cr, etc., were within the normal range (Fig. 8e). In addition, we examined the Fe2+ levels in the heart, liver, and kidney tissues of the mice and found that there was no significant difference between the heart, liver, and kidney tissues of the ACE-treated mice and those of the control group (Fig. 8f). These results indicate that ACE at therapeutic doses has no significant risk of ferroptosis induction in normal tissues and that the clinical application of ACE provides a solid basis for translational medicine.
Fig. 8
Comments (0)