
Remember me

The primary objective of this study was to investigate the therapeutic and preventive effects of miRNA 15-29148 antagomir targeting chondrocytes in CIA mice. To assess the therapeutic efficacy, normal mice and surgically induced RA models were randomly divided into seven groups: Normal, CIA, NP, NP/agomir control, NP/miRNA 15-29148 agomir, NP/antagomir control, and NP/miRNA 15-29148 antagomir, with each group comprising eight animals. The treatment was administered via intra-articular injection at a dose of 10 µL every 5 days for 80 days, and disease severity was evaluated at the end of this period. To evaluate the preventive effects, normal mice were randomized into three groups: Normal, CIA, and NP/miRNA 15-29148 antagomir. Prophylactic administration was conducted for 6 weeks before inducing the CIA model, with each group also comprising eight animals. The drug was administered intra-articularly at a dose of 10 µL once every 7 days for a total of 8 weeks, and disease severity was assessed 21 days post-modeling. Specific experimental procedures and additional methodological details are provided in the Detailed Experimental Methods section.
Ethics statementAnimal experiments were performed in accordance with the “Guide for Laboratory Animals” of Jiangnan University and received approval from the Laboratory Animal Ethics Committee of Jiangnan University, Wuxi, China (Approval No. JN. No20240115d0480520[047]). Informed consent was obtained from all patients involved in the study, and the research protocol was approved by the Research Ethics Committee of Anhui Medical University, Hefei, China (Approval No. S20210085).
Inclusion and exclusion criteriaPatients with RA were diagnosed according to the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for RA and were classified based on the Disease Activity Score 28 (DAS28). The inclusion criteria for patients with RA were age >18 years and a disease duration of at least 6 weeks. The exclusion criteria were as follows: (1) a known diagnosis of autoimmune diseases other than RA, (2) a history of severe chronic infections or any current infection, (3) a diagnosis of cancer, (4) shift work, (5) pregnancy and breastfeeding, and (6) receipt of antibiotic treatment within 1 month before participation in this study. For normal/osteoarthritis (OA) controls, individuals had to meet the following inclusion criteria: age >18 years and recent average liver and kidney function screening values. The exclusion criteria were as follows: (1) a known diagnosis of autoimmune diseases, (2) a history of severe chronic infections or any current infection, (3) a diagnosis of cancer, (4) shift work, (5) pregnancy and breastfeeding, and (6) receipt of antibiotic treatment within 1 month before participation in this study.
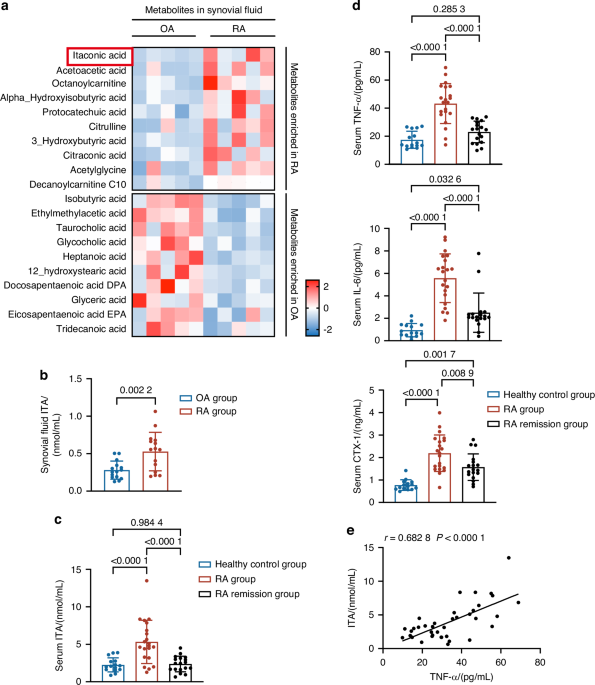
Human specimen collectionKnee joint effusions from patients with RA were examined using ultrasound, and synovial fluid was extracted. Control synovial fluid samples were obtained from patients with OA. The exclusion criteria included the presence of cancer, infections, and autoimmune diseases. Synovial and cartilage tissues from patients with RA were collected during arthroplasty procedures, with the exclusion criteria being cancer, infections, and autoimmune diseases, excluding RA. Normal synovial and cartilage tissues were obtained from individuals who underwent amputation due to a car accident or from patients with OA undergoing arthroplasty. The exclusion criteria for these individuals included the presence of cancer, infections, and autoimmune diseases.
Isolation and passage of human primary synovial fibroblastsSynovial tissues obtained from knee replacement surgeries of patients with RA or OA were transported to a laminar flow hood within 30 min of collection. The tissues were washed 3–5 times with 75% ethanol and sterile phosphate-buffered saline (PBS) (RG-CE-10, Ketu Biotech). Residual fat and other extraneous tissues were meticulously removed from the synovial tissues using scissors. The cleaned tissues were then placed in Dulbecco’s modified Eagle’s medium (DMEM) high-glucose medium (Gibco) containing 20% fetal bovine serum (FBS; Gibco). The synovial tissues were cut into approximately 25 mm² pieces using sterile ophthalmic scissors and forceps. These pieces were transferred to the bottom of a sterile culture flask with a sterile Pasteur pipette. The culture flasks were inverted, and an appropriate volume of DMEM high-glucose medium with 20% FBS was added. The inverted flasks were carefully placed in an incubator and left undisturbed for 6 h to allow the tissue pieces to adhere to the flask surface. After tissue adherence, the flasks were gently turned upright and placed back in the incubator. The culture medium was not changed for the first 3 days. Starting from day 4, the medium was changed every 3 days until the density of synovial fibroblasts migrating outward from the tissue pieces exceeded 70%. Subsequently, the cells were passaged using trypsin digestion. Human primary synovial fibroblasts from passages 3 to 6 were used for in vitro experiments.
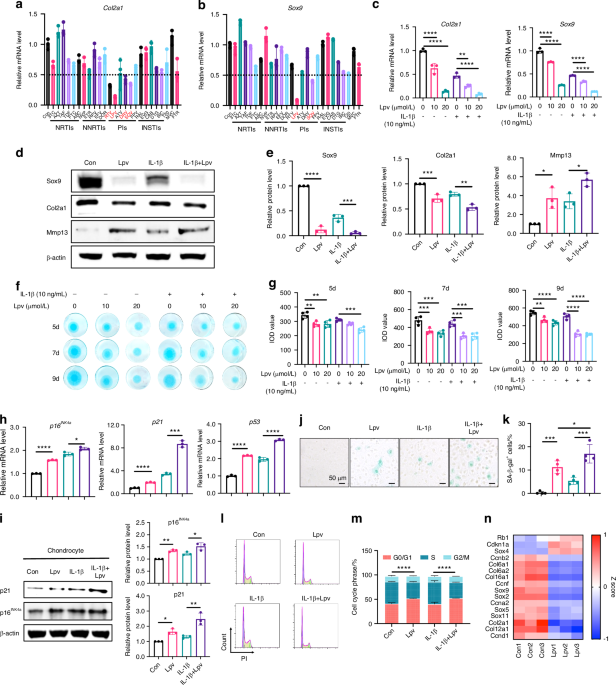
Isolation and passage of human primary chondrocytesCartilage tissues obtained from knee replacement surgeries of patients with RA or OA were promptly transported to a laminar flow hood within 30 min. The tissues were thoroughly washed 3–5 times with 75% ethanol and sterile PBS. Using sterile ophthalmic forceps and scissors, the cartilage tissues were cut into approximately 4 mm² pieces. The tissue pieces were softened by treating them with trypsin at 37 °C for 30 min, with intermittent shaking every 5 min. Following this softening step, the cartilage tissues were digested with 0.3% type II collagenase (Sigma) prepared in DMEM high-glucose medium without FBS for 8–12 h. The resulting cell suspension was then filtered through a sterile 75 μm cell strainer (NEST). The single-cell suspension was centrifuged and subsequently cultured in DMEM high-glucose medium (RG-CE-2, Ketu Biotech) containing 10% FBS. Any undigested tissue pieces were subjected to additional digestion if necessary. Once the cell confluence exceeded 70%, the cells were passaged using trypsin (Gibco) digestion. Human primary chondrocytes from passages 3 to 5 were used for in vitro experiments.
Identification of primary human chondrocytesThird-generation primary human chondrocytes were seeded into 6-well plates (Corning). When the cells reached 70%–80% confluence, the medium was discarded, and the cells were washed three times with PBS. The cells were subsequently fixed with 4% paraformaldehyde (Biosharp) for 30 min. Following fixation, the chondrocytes were stained using Toluidine Blue (Solarbio) or subjected to type II collagen immunohistochemistry. The positive rate of the cells was then observed under a microscope.
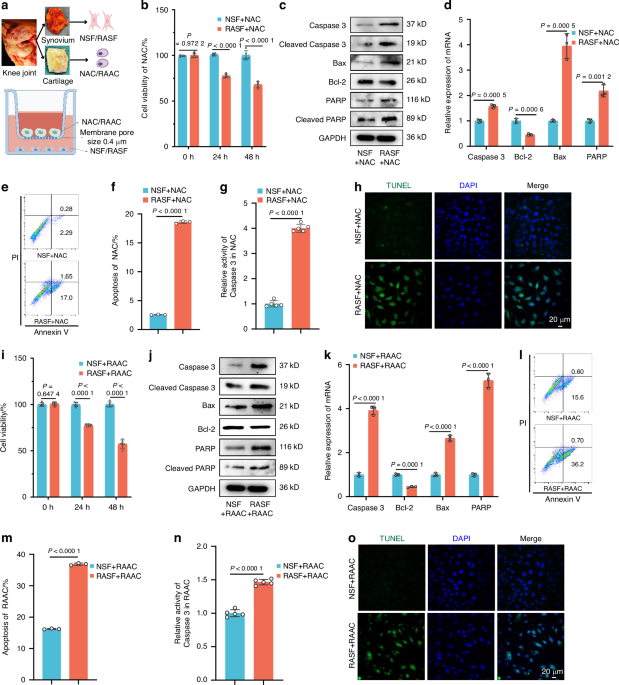
Co-culture of primary synovial fibroblasts and primary chondrocytesPrimary synovial fibroblasts and primary chondrocytes from passages 3-5, in their logarithmic growth phase, were selected for co-culture. The cells were washed with PBS and digested with 0.25% trypsin for further use. A transwell plate (Corning) with a pore size of 0.4 μm was employed for the co-culture. Synovial fibroblasts (donor cells) were seeded in the lower chamber, while chondrocytes (recipient cells) were seeded in the upper chamber.
Cell viability assayCell viability was assessed by seeding cells into a 96-well culture plate. The Cell Counting Kit-8 (CCK-8; Sigma-Aldrich) was used to measure viability, which involves the use of 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt. Absorbance was recorded using a microplate reader (Thermo Scientific, Waltham, MA, USA).
Annexin V-FITC/PI dual staining for apoptosis detectionCells were digested with 0.25% trypsin without EDTA, and the digestion was stopped by adding culture medium. The cell suspension was centrifuged at 2 000 r/min for 8 minutes. After centrifugation, the supernatant was discarded, and 1× Annexin V binding buffer was added for another round of centrifugation. The supernatant was discarded again, and 300 μL of 1× Annexin V binding buffer was added to each tube. The cells were resuspended using a pipette and transferred to flow cytometry tubes. In a dark environment, 5 μL of FITC dye (BD Biosciences) was added to the cell suspension and incubated for 15 minutes. Subsequently, 6.5 μL of PI dye (BD Biosciences) was added and incubated for 5 minutes. After incubation, 200 μL of 1× Annexin V binding buffer was added to each tube to bring the total volume to 500 μL. Apoptosis levels were then detected using a flow cytometer (Beckman Coulter, CA, USA).
TUNEL staining for apoptosis detectionCells intended for apoptosis analysis were suspended in PBS buffer at a concentration of 2 × 107 cells/mL. A volume of 50–100 μL of cell suspension was dropped onto poly-L-lysine-coated slides and gently spread using a clean slide after cell adhesion. The cells were fixed with 4% paraformaldehyde solution for 25 minutes. After fixation, the slides were washed twice with PBS for 5 minutes each time, and excess PBS was removed. Cells were permeabilized with 0.2% Triton X-100 solution in PBS at room temperature for 5 minutes. Subsequently, the slides were washed three times with PBS for 5 minutes each. Excess PBS was removed, and 100 μL of 1× Equilibration Buffer was added. The slides were then incubated at room temperature for 30 minutes. The Equilibration Buffer was removed, and 50 μL of TdT reaction buffer (abcam) was added to the slides, followed by incubation at 37 °C for 60 minutes. After incubation, the slides were washed three times with PBS for 5 minutes each in a dark environment. After the final wash, the slides were washed three times with PBS containing 0.1% Triton X-100 and 5 mg/mL BSA to reduce cellular background. Subsequently, 2 μg/mL DAPI solution (Thermo Scientific) was added and incubated for 5 minutes. After three additional washes with PBS, the slides were immediately observed under a confocal laser scanning microscope to visualize green and blue fluorescence.
Caspase-3 activity analysisCaspase-3 activity was assessed using the Caspase-3 Colorimetric Assay Kit (R&D Systems) following the manufacturer’s protocol.
Western BlottingProtein lysates from tissues or cells were prepared using RIPA buffer supplemented with protease and phosphatase inhibitors. The protein concentration was determined using the BCA Protein Assay Kit (ThermoFisher Scientific). Samples were separated by 10% SDS-PAGE and transferred onto PVDF membranes. The membranes were then incubated overnight at 4°C with specific primary antibodies (refer to Table S1) in TBS-T buffer containing 5% BSA. After washing with TBS-T, the membranes were incubated with HRP-conjugated anti-rabbit IgG or anti-mouse IgG antibodies for 2 h. Immunocomplexes were visualized using an ECL detection kit (ThermoFisher Scientific) and detected by chemiluminescence.
Total RNA isolation, cDNA synthesis, and RT-qPCRTotal RNA was isolated from tissues or cells using TRIzol reagent (Thermo Scientific, Waltham, MA, USA). The quantity and quality of mRNA were assessed using a nanodrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). cDNA was synthesized with the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems, Foster City, CA). For mRNA analysis, RT-qPCR was performed using specific primers and SYBR Green PCR Master Mix (Applied Biosystems) on a QuantStudio 12k Flex System (Applied Biosystems, Foster City, CA). GAPDH was used as a normalization control. The results were analyzed using the comparative Ct (ΔΔCt) method (2−ΔΔCt for logarithmic transformation). For miRNA analysis, reverse transcription and real-time PCR were conducted according to protocols specified by GenePharma (Shanghai, China). U6 was used as the normalization control. All reactions were run on a QuantStudio 12k Flex System (Applied Biosystems, Foster City, CA) and analyzed using the comparative Ct (ΔΔCt) method (2−ΔΔCt for logarithmic transformation). Refer to Table S2 for corresponding primer sequences.
sEV extraction and characterizationsEV were isolated from cell culture supernatants using differential ultracentrifugation. Briefly, cells were cultured in T75 culture flasks for 48 h to extract sEV. After culturing, the cell culture supernatant was collected and centrifuged at 330 g for 10 minutes to remove residual dead cells. The supernatant was transferred to new centrifuge tubes and centrifuged at 3 000 g for 30 minutes to remove cell debris. Subsequently, the supernatant was transferred to clean ultracentrifuge tubes and centrifuged at 10 000 g for 60 minutes to remove larger vesicles. Following centrifugation, the supernatant was filtered through a 0.22 μm filter and transferred to new ultracentrifuge tubes for ultracentrifugation at 120 000 g (Beckman Coulter) for 120 minutes to pellet sEV. After extraction, sEV proteins were lysed directly using a cell lysis buffer, and sEV RNA was extracted using Trizol. sEV were suspended in PBS, collected in enzyme-free centrifuge tubes, and stored at -80°C for up to 7 days. Total sEV protein was quantified using the BCA Protein Assay Kit (ThermoFisher Scientific). sEV identification was performed using several methods: Western blotting to investigate the expression of sEV positive proteins (CD9, CD63, Flotillin-1) and sEV negative proteins (Calnexin, β-actin); nanoparticle tracking analysis to analyze the size distribution of extracted sEV, typically ranging from 30 to 200 nm; and transmission electron microscopy to observe the typical bilayer membrane structure and confirm sizes between 30 and 200 nm. These methods collectively confirmed the standard characterization of the extracted sEV.
sEV labeling and internalizationPKH26 dye (Sigma-Aldrich) was used to label sEV for tracking and internalization studies. Initially, 80 μL of PKH26 dye at a 1 mmol/L concentration was diluted in 10 mL of Diluent C solution to achieve a final concentration of 8 μmol/L (Solution 1). Subsequently, 1 mg of sEV dissolved in 2 mL of DPBS (Dulbecco’s Phosphate-Buffered Saline) was mixed with 8 ml of Diluent C to obtain Solution 2. Solutions 1 and 2 were combined and gently vortexed with a pipette, followed by incubation at 4 °C for 5 minutes. After incubation, 10 mL of DMEM culture medium containing 10% sEV-free serum was added to bind excess dye. The sEV were then diluted with 100 mL of DPBS and transferred to ultracentrifuge tubes. Using a swing-bucket rotor, the sEV were centrifuged at 120 000 × g at 4 °C for 120 minutes. After centrifugation, the supernatant was discarded, and the sEV were resuspended in an appropriate volume of DPBS (Gibco) and washed twice in the centrifuge tube to completely resuspend the dyed sEV. The dyed sEV were filtered through a 0.22 μm filter and added to the culture medium of chondrocytes. Once the sEV were internalized by the chondrocytes, the cells were washed three times with pre-warmed PBS at 37°C to remove the culture medium. To remove any non-internalized sEV, an appropriate amount of green-labeled cholera toxin subunit B was added and incubated for 10 minutes. After incubation, the cells were washed three times with pre-warmed PBS buffer and incubated with DAPI for 10–15 minutes. The cells were then washed three times with PBS and observed using a confocal laser scanning microscope (Zeiss LSM 780) to examine the intensity and distribution of red, green, and blue fluorescence.
MicroRNA microarray analysis of sEVHigh-purity sEV were isolated using differential ultracentrifugation as described previously. Total RNA was extracted from the sEV using TRIzol according to the manufacturer’s instructions. The quality of RNA samples was assessed using a 5300 Bioanalyzer (Agilent), and the quantity was determined with an ND-2000 spectrophotometer (NanoDrop Technologies). Only RNA samples meeting high-quality criteria (OD260/280 = 1.8–2.2, OD260/230 ≥ 2.0, RIN ≥ 6.5, 28S:18S ≥ 1.0, >1 μg) were selected for library construction. RNA purification, reverse transcription, library construction, and sequencing were performed by Majorbio Bio-Pharma Technology Co., Ltd. (Shanghai, China), following Illumina’s recommendations. For each sample, 1 μg of total RNA was used to prepare small RNA libraries using the QIAseq miRNA Library Kit (Qiagen). Activated 5’ and 3’ adapters were ligated to the total RNA, followed by first-strand cDNA synthesis using reverse transcriptase and random primers. PCR amplification was conducted for 11-12 cycles, and the appropriate-sized fragments were separated on a 6% Novex TBE PAGE gel. After quantification with Qubit 4.0, single-end RNA-seq libraries were sequenced using an Illumina NovaSeq Xplus sequencer. Raw reads in fastq format were processed with fastx tools to remove 3’ adapters, poly-N segments, 3’ low-quality bases (Sanger base quality < 20), and sequencing adapters, resulting in clean reads. Identical sequences ranging from 18 to 32 nucleotides were counted and removed from the initial dataset. Bowtie software was used to annotate chromosomal positions according to the reference genome data. Known miRNAs were identified by mapping small RNA tags to the miRBase 22.0 database. Remaining tags were aligned with Rfam and Repbase databases to exclude non-coding RNAs and repeat sequences. Unannotated tags were predicted as novel miRNAs using mirdeep2 based on genomic positions and hairpin structures. The expression levels of each miRNA were calculated using the TPM (transcripts per million reads) method. Differential expression analysis was conducted using DESeq2 or DEGseq, considering |log2FC| ≥ 1 and FDR ≤ 0.05 (DESeq2) or FDR ≤ 0.001 (DEGseq) as criteria for significantly differentially expressed miRNAs. Target gene prediction of miRNAs was performed using miRanda, and predicted target genes were annotated using the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Functional enrichment analysis, including GO and KEGG pathway analyses, was conducted to identify significantly enriched pathways and GO terms compared to the whole genome background, with a significance level of P-adjust ≤ 0.05. Additionally, functional enrichment analysis for graphene oxide was performed using Goatools, and KEGG pathway analysis was conducted using KOBAS.
Transient transfectionFor transient transfection experiments, 200 μL of Opti-MEM medium (Gibco) was added to two sterile 1.5 mL centrifuge tubes labeled as Tube A and Tube B. In Tube A, transfection reagent was added, while in Tube B, the transfection product (miRNA mimics/inhibitors) (GenePharma) was added and allowed to disperse for 5 minutes to ensure thorough mixing. Subsequently, the dispersed transfection product from Tube B was slowly added to the dispersed transfection reagent in Tube A, and the two components were thoroughly mixed for 30 minutes. Within 60 minutes, the mixed solution was slowly and dropwise added to the cells to be transfected, in 2 mL of DMEM high-glucose medium containing FBS, which was added beforehand. For RNA-based experiments, transfection was conducted for 24 h; for protein-based experiments, transfection continued for 72 h. GP-transfect-Mate transfection reagent (GenePharma) was used, with 6.5 μL used per primary human chondrocyte. The miRNA mimics were used in amounts of 9 μL, and the inhibitors of miRNA were used in amounts of 18 μL. Refer to Table S3 for corresponding sequences.
Luciferase reporter assayCells were seeded into a 24-well plate and co-transfected with firefly luciferase reporter constructs encoding wild-type CIAPIN1 3′-UTRs (CIAPIN1-3′-UTR-WT) or mutated CIAPIN1 3′-UTR regions (CIAPIN1-3′-UTR-Mut) (GenePharma), along with miRNA 15-29148 mimics or control mimics, using Lipofectamine 3000 (Invitrogen). Firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay Kit (Beyotime) according to the manufacturer’s instructions. All experiments were performed in triplicate to ensure statistical reliability.
MiceMale DBA/1J WT mice, aged 8-10 weeks, were obtained from Nanjing JiCui Biotechnology Co., Ltd. (Nanjing, China). All mice were housed under specific pathogen-free conditions with a 12:12-h light/dark cycle. The mice were utilized for collagen-induced arthritis (CIA) and complete/incomplete Freund’s adjuvant-induced arthritis experiments.
Induction of Collagen-Induced Arthritis (CIA) ModelCollagen-induced arthritis (CIA) was induced in DBA/1J mice on days 1 and 21. Briefly, 100 μg of bovine type II collagen (Chondrex, Redmond, WA, USA) emulsified in an equal volume of complete Freund’s adjuvant (Chondrex) was subcutaneously injected at the base of the tail. On day 21, a second immunization was given with 100 μg of bovine type II collagen emulsified in an equal volume of incomplete Freund’s adjuvant (Chondrex). Following the second immunization, arthritis severity was assessed using the following scoring criteria: 0, no evidence of erythema and swelling; 1, erythema and mild swelling confined to the digits; 2, erythema and mild swelling extending from the ankle to the midfoot (tarsals); 3, moderate swelling and erythema extending from the ankle to the entire foot (metatarsals); 4, severe erythema and swelling encompassing the ankle, foot, and digits, resulting in ankylosis and/or deformity. A clinical score (0-16) was calculated by summing the scores from all limbs, assessed every 2 days. Clinical evaluations were performed in a blinded manner, and arthritis scores were analyzed using a two-way analysis of variance.
Treatment of CIA MiceDBA/1J mice with established collagen-induced arthritis (CIA) were randomly divided into 6 groups, each consisting of 8 mice. An additional 8 normal DBA/1J mice from the same batch were included as healthy controls, resulting in a total of 7 groups comprising 56 mice. Thirty-five days after initial immunization, CIA mice received 12 intra-articular injections into the joint cavity, administered every 5 days. The treatment groups were as follows: Healthy control group (no treatment), Saline (10 μL, PBS), NPs (5 mg/mL, 10 μL), NPs/agomir control (weight ratio 16:1, 10 μL), NPs/miRNA 15-29148 agomir (weight ratio 16:1, 10 μL), NPs/antagomir control (weight ratio 16:1, 10 μL), NPs / miRNA 15-29148 antagomir (weight ratio 16:1, 10 μL). During the treatment period, clinical parameters of CIA mice were monitored. At the end of the treatment, hind paw thickness was measured using a caliper, and RA-related behavioral assays were conducted.
Prevention of CIA in MiceMale DBA/1J WT mice aged 8-10 weeks were randomly divided into 3 groups, each consisting of 8 mice. One group served as the normal control group and did not receive any treatment or CIA induction. The other two groups received intra-articular injections of saline (10 μL) and NPs / miRNA 15-29148 antagomir (weight ratio 16:1, 10 μL) once a week, for a total of 6 times. After the fifth administration, these two groups of mice were subjected to initial CIA induction. After 21 days of initial induction, a booster immunization was performed. The experiment concluded 40 days after the booster immunization. During the process, the clinical parameters of CIA mice were monitored. At the end of the experiment, hind paw thickness was measured using a caliper, and RA-related behavioral assays were conducted.
Beam walking testA narrow beam measuring 3.0 cm wide and 75 cm long was positioned 25 cm above a platform. Prior to receiving treatments, all animals underwent training to walk on the beam. Following treatment, the time each animal required to traverse the beam was measured using a stopwatch. Three consecutive trials were conducted, and the average of these three values was calculated as the walking time for each mouse.
Footprint assayThe plantar surface of each mouse’s hind paw was coated with black ink. Subsequently, the mouse freely walked along a corridor measuring 1 m in length and 10 cm in width, with white paper placed at the bottom. The distance between consecutive hind paw prints was measured using a digital caliper.
Micro-computed Tomography (micro-CT) analysisAt the conclusion of the treatment period, mice were euthanized, and their hind paws were fixed in 4% (w/v) paraformaldehyde (RG-GD-01, Ketu Biotech) for 24 h. Subsequently, the hind paws were scanned using a micro-CT system with a 9 μm isotropic resolution on Quantum GXμ-CT (PerkinElmer, Ontario, Canada). The acquired images were then subjected to three-dimensional reconstruction and segmentation using MATERIALIZE MIMICS 19.0 software (Materialize, Leuven, Belgium).
Measurement of serum inflammatory factorsAt the conclusion of the animal experiment, whole blood was collected from CIA mice. The blood samples were allowed to clot at room temperature (25 °C) for 30 minutes, followed by centrifugation at 1 500 g for 30 minutes to separate the serum. The upper serum layer was carefully collected for further analysis. The expression levels of TNF, IL-1β, and IL-6 in the serum were measured using ELISA kits specific for each cytokine, according to the manufacturer’s instructions (R&D Systems).
Histopathological analysis of joint tissuesAnkle joint tissues from the arthritis model were collected and fixed in 4% (w/v) paraformaldehyde at 4°C for 24 h. Decalcification was performed using 10% (w/v) EDTA (RG-TG-01, Ketu Biotech) solution at room temperature (25°C) for 3 weeks. After decalcification, the joints were embedded in paraffin, sectioned into 30 μm thick slices, and stained with hematoxylin and eosin (H&E) for general histological analysis. For cartilage evaluation, sections were stained with Safranin O/Fast Green. The stained sections were observed and photographed using an Olympus DP70 microscope (Tokyo, Japan).
Fluorescence in situ Hybridization (FISH) and Immunohistochemistry (IHC)Paraffin-embedded tissue sections were deparaffinized and permeabilized with proteinase K at 37 °C for 10 minutes. Endogenous peroxidase activity was blocked with 3% (vol/vol) H2O2, followed by dehydration and rehydration steps. For FISH, hybridization was performed using a 40 nmol/L double-DIG LNA™ microRNA probe specific for miRNA 15-29148 (Exiqon, Vedbaek, Denmark) at 52°C for 1 h. After washing with 5x SSC buffer, 1x SSC buffer, and 0.2x SSC buffer, the sections were blocked for 15 minutes and incubated with anti-DIG-POD antibody (Roche Applied Science, Indianapolis, IN, USA). Fluorescence detection was carried out using the TSA Plus Fluorescence System (PerkinElmer, Waltham, Massachusetts, USA) according to the manufacturer’s instructions. For IHC, paraffin-embedded tissue sections were deparaffinized in xylene, rehydrated through a graded alcohol series, and rinsed in water. The sections were then incubated with primary antibodies (see Table S1) at room temperature for 1 h, followed by incubation with biotinylated secondary antibodies (see Table S1) for 30 minutes. The staining was visualized using the Vectastain ABC Kit and DAB Peroxidase Substrate Kit (Vector Laboratories).
Biocompatibility analysisUpon completion of the animal experiments, blood and major organs were collected from the mice. The serum was separated using the methods described above, and concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), creatinine (CREA), and blood urea nitrogen (BUN) were quantified using an Olympus AU400 fully automated clinical chemistry analyzer. Each organ (heart, liver, spleen, lungs, kidneys) was fixed in 4% (w/v) paraformaldehyde and embedded in paraffin. Sections of 5 μm thickness were prepared from each organ and stained with hematoxylin and eosin (H&E) to detect any pathological changes.
Immunofluorescence stainingParaffin-embedded tissue sections were deparaffinized by heating in a 60 °C oven for 15 minutes and then hydrated through a series of xylene and graded alcohols. For antigen retrieval, heat-induced epitope retrieval was performed using a vegetable steamer: slides were incubated in 10 mmol/L citrate buffer at 60°C for 40 minutes followed by washing in PBS. At room temperature, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 minutes. Subsequently, the appropriate primary antibodies (refer to Table S1) were applied and incubated overnight. The following day, corresponding fluorescent secondary antibodies (refer to Table S1) and DAPI were applied, and the samples were observed using laser confocal microscopy (Zeiss LSM 780).
Synthesis and characterization of nanoparticles (NPs)Forty milligrams of G5.0 PAMAM (Dendritech, Inc, MI, USA) were dissolved in 2 mL of 0.1 mol/L pH 7.4 PBS. Subsequently, 3.0 mg of Cy5.5 NHS ester (Lumiprobe Corporation, Hallandale Beach, FL), dissolved in 100 μL of DMSO, was added to the solution, which was vigorously stirred at room temperature for 24 h. Excess Cy5.5 was removed by ultrafiltration using an Amicon Ultra-15 centrifugal filter unit (MWCO 3000, Millipore, Billerica, MA). Cy5.5-PAMAM was then conjugated with NHS-PEG2000-MAL (Laysan) at a 1:6 ratio in phosphate buffer solution (pH 8.0) at room temperature for 12 h. The resulting conjugate, Cy5.5-PAMAM-PEG-MAL, was purified by ultrafiltration using a 14 kD molecular weight cut-off membrane and freeze-dried for storage at 4 °C. For the next step, 1.2 µmol of Cy5.5-PAMAM-PEG-MAL was dissolved in 5 mL of nuclease-free phosphate buffer solution (pH 7.0) and reacted with 0.6 µmol of SH-tgg2 at 4 °C for 12 h. The resulting product, Cy5.5-PAMAM-PEG-tgg2, was purified by ultrafiltration and stored at 4 °C. For nanoparticle (NPs) preparation, miRNA (15 µL, 1.5 µg) was mixed with tgg2-PEG-PAMAM-Cy5.5 (5 mg/mL) at different mass ratios (2:1, 4:1, 8:1, 16:1, 32:1, and 64:1). The mixture was gently vortexed for 30 seconds and then allowed to stand for 30 minutes to prepare stable tgg2-PEG-PAMAM-Cy5.5/miRNA 15-29148 agomir/miRNA 15-29148 antagomir NPs. Gel retardation assay was performed to determine the optimal dose of tgg2-PEG-PAMAM-Cy5.5 required for complete encapsulation of miRNA. Characterization of NPs was conducted using nuclear magnetic resonance spectroscopy (1H NMR), dynamic light scattering (DLS), and transmission electron microscopy (TEM).
Gel Retardation AssayNanoparticles (NPs) were prepared by mixing miRNA 15-29148 agomir/miRNA 15-29148 antagomir with NPs at different N/P ratios. After incubation at 37°C for 30 minutes, gel retardation analysis was performed using 1% (w/v) agarose gel electrophoresis (120 V, 20 min). Bands were visualized using the Universal Hood II system (Bio-Rad, USA), with a 10 000 bp DNA ladder (Takara, Dalian, China) used as a molecular weight marker. Uncropped gel scans were provided as source data files.
Cellular uptake and intracellular mechanism analysis of NPsChondrocytes were treated with PEG-PAMAM-Cy5.5 or tgg2-PEG-PAMAM-Cy5.5 and incubated at 37 °C for 1–8 h. For one set of cells, after 2, 4 and 8 h of incubation, they were fixed with 4% paraformaldehyde for 30 minutes, stained with DAPI, and observed under a laser confocal microscope (Carl Zeiss Microscopy LLC, Jena, Germany) to assess NP uptake at different time points. For another set of cells, after incubation for 1, 3 and 6 h, they were further incubated at 37°C for 1 h in cell culture medium containing 50 nmol/L DAPI and 100 nmol/L LysoTracker Green. After incubation, cells were fixed with 4% paraformaldehyde and observed under a laser confocal microscope (Carl Zeiss Microscopy LLC) to evaluate the intracellular distribution of NPs at different time points.
In vivo targeting analysis of NPsMale DBA/1J WT mice aged 8-10 weeks were intra-articularly injected with Free Cy5.5, PAMAM-Cy5.5, or tgg2-PAMAM-Cy5.5 at a dose of 5 mg/mL, 10 μL. Mice were euthanized on days 1, 3, 5 and 10, and their organs and limbs were dissected. Tissues were examined using the CALIPER IVIS Lumina III in vivo imaging system (PerkinElmer, USA) to detect tissue fluorescence (λex: 640 nm, λem: 680 nm).
Statistical analysisData are presented as the mean ± SD of three independent experiments. Differences between two groups were determined using the Wilcoxon rank-sum test or Student’s t-test, while variance analysis (ANOVA) or the Kruskal–Wallis test was used for comparisons among three groups, based on the data type. Spearman correlation was used to assess the correlation between clinical information and the differential abundance of taxa. All graphs and statistical analyses were performed using GraphPad Prism 9 (GraphPad Software Inc., La Jolla, CA, USA).
Comments (0)