Animal studies
All animal studies were performed under protocols approved by the Western Sydney Local Health District Animal Ethics Committee (no. 4291 and no. 4369) and in accordance with the Australian code for the care and use of animals for scientific purposes developed by the National Health and Medical Research Council of Australia. Cd47−/− mice (B6.129S7-Cd47tm1Fpl/J, back-crossed to C57BL/6 mice for 15 generations, https://www.jax.org/strain/003173#) and 3- to 4-month old male C57BL/6 (Cd47+/+, WT) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and bred at the Australia Bio Resources (Moss Vale, NSW, Australia). For experimental work, male mice were housed in the Westmead Institute for Medical Research Animal Research Facility with ad libitum access to regular chow (8% energy from fat; Gordon’s Specialty Stockfeeds, Yanderra, Australia) and water.
Human plasma and tissue samples
Healthy individuals or individuals with type 1 diabetes and normal renal function (based on eGFR determined by CKD-EPI) were recruited from outpatient clinics at Westmead Hospital. Blood was collected in K2-EDTA tubes without a tourniquet using a 23-gauge needle, and placed immediately on ice. Platelet-poor plasma was generated by centrifugation at 2500 g for 15 min at 4°C without brake, then stored at −80°C until analysis. Plasma TSP1 concentration was determined by ELISA (Abcam, Cambridge, UK). The study was approved by the Human Research Ethics Committee of Western Sydney Local Health District (HREC LNR/12/WMEAD/114 and LNRSSA/12/WMEAD/117). All participants provided written consent.
Human pancreas samples used in this study were from deceased donors whose organs were consented for research and unsuitable for either whole-organ or islet transplantation. This study was approved by the Human Research Ethics Committee of Western Sydney Local Health District (HREC 2021/PID02353). All families of organ donors provided written informed consent. Donor details are listed in electronic supplementary material (ESM) Table 1.
Primary murine islet isolation
Mouse islet isolation was performed as described previously [17] and outlined in detail in ESM Methods. Islets were stained with Dithizone dye (Cole-Parmer Scientific Experts, Vernon Hills, IL, USA) before counting and use in experimental work.
Human islet isolation
Research-consented human islets unsuitable for clinical transplantation were isolated following the standard protocol carried out by the National Islet Transplantation Consortium at Westmead Hospital [18]. Donor details are provided in the human islet checklist (see ESM).
Islet exogenous stressor experiments
Murine islets, human islets or MIN6 cells (no. C0018008; AddexBio, San Diego, CA, USA) were exposed to normoxia (fraction of inspired oxygen [FIO2] 21%) or hypoxia (FIO2 1%) for 24 h and then stimulated with basal (2.8 mmol/l) or high (16.7 mmol/l) glucose for 1 h. In separate experiments, cells were supplied with high-glucose media and treated with 0.5 µmol/l thapsigargin (Sigma-Aldrich) for 18 h. The MIN6 cell line (passages 22–30) was tested monthly for mycoplasma and no contamination was identified.
Limiting CD47 signalling via RNA interference or antibody blockade
Specific to these studies, mouse Cd47 and control siRNA (Ambion, Life Technologies, Austin, TX, USA) and human CD47 and control siRNA (ThermoFisher Scientific, Waltham, MA, USA) were used as published previously [16]. In brief, MIN6 cells or human islets were seeded onto 6 cm petri dishes, serum-starved for 1 h, then were treated with either CD47-targeted or scrambled control siRNA according to the manufacturer’s instructions (Ambion, Protocol Pub. No. MAN0007836 Rev. 1.0) for 48 h. For antibody blockade, cells were supplied with DMEM high-glucose media and treated with IgG isotype control or anti-CD47 antibody (1 μg/ml) for 15 min prior to experiment initiation.
Insulin secretion measurements
Following respective cell culture experiments, supernatant fractions were collected and any residual cells were removed from the media by centrifugation at 200 g for 4 min at 4°C. Samples were frozen at −80°C until insulin secretion was determined in bulk by ELISA according to the manufacturer’s instructions (Ultra-sensitive Mouse Insulin ELISA no. 90080; Crystal Chem, Elk Grove Village, IL, USA).
Cell viability, senescence and LDH assays
Cell viability was measured using an XTT Cell Viability Kit (no. 9095, Cell Signalling Technology). Briefly, 104 WT or CD47−/− islets/well were seeded into a 96-well microplate and viability was determined at 450 nm using Proteomics SpectraMax iD5 Plate Reader (VWR International, Radnor, PA, USA).
Senescence was assessed with a Mammalian β-Galactosidase Assay Kit (no. 9860, ThermoFisher Scientific). MIN6 cells were treated with control scramble or CD47 siRNA according to the protocol previously described [16]. Cells were then exposed to hypoxia, supplied with β-galactosidase staining solution and incubated at 37°C overnight. Assessment was performed under light microscopy, and the per cent area of blue dye staining from at least five areas per well per group were evaluated using ImageJ (https://imagej.net/ij/; last accessed 18 Dec 2024).
Lactate dehydrogenase (LDH) from cell culture supernatant fractions was measured using the Lactate Dehydrogenase Assay Kit (ab102526, Abcam) according to the manufacturer’s instructions. In brief, supernatant fractions were stored at −80°C, thawed simultaneously and brought to room temperature. The NADH standard, dilutions and samples were run in duplicate, and the plate was read initially at 450 nm (Proteomics SpectraMax iD5 Plate Reader). Absorbance was read a second time after incubating the plate for 30 min at 37°C, and this reading was subtracted from the initial reading. LDH activity (mU/ml) was determined via the formula: (B × sample dilution) / (30 min × pre-treated sample volume), where B is the difference in absorbance reading. Corrected absorbance values were calculated to generate a standard curve and LDH concentration in samples was determined using the formulaic calculation provided.
Seahorse assay
To assess metabolic responses in cells, Agilent Seahorse XF Cell Mito Stress Test and XFe24 Extracellular Flux Assay Kit (Agilent Technologies, Santa Clara, CA, USA) assays were used. MIN6 cells were seeded at 105 cells/well in a Seahorse assay plate and treated with CD47 siRNA for 48 h. A day prior to running the assay, a sensor cartridge plate was hydrated by supplying Seahorse calibrant to each well and incubating it overnight in a non-CO2 incubator. On the day of the assay, compounds were resuspended in freshly prepared Seahorse assay medium, diluted in the assay medium to working concentrations (1 µmol/l oligomycin, 0.8 µmol/l FCCP and 0.5 µmol/l rotenone/antimycin A) and injected into respective ports (port A, oligomycin; port B, FCCP; port C, rotenone/antimycin A; and port D, empty). Medium in the plate was changed to Seahorse assay medium and the cells were incubated for 1 h in a non-CO2 incubator. The Seahorse assay was then run according to pre-set cycles designed on the Wave app (5 measurement cycles for all compounds) and analysed.
Immunofluorescence and morphometry on tissue sections and cells
Human pancreas, as well as mouse and human islets embedded in Optimal Cutting Temperature compound (OCT) medium, were sectioned at 7 µm thickness, fixed in 4% (vol./vol.) paraformaldehyde (PFA), washed with PBS and permeabilised with 0.2% Triton (Sigma-Aldrich). Sections were then blocked in 2% BSA and incubated overnight at 4°C with the relevant primary antibodies (listed in ESM Table 2). Sections were then probed with respective fluorophore-tagged secondary antibodies (ESM Table 3) for 1 h at room temperature, stained with DAPI (1 µg/ml; Sigma-Aldrich), mounted (Dako, Agilent, Denmark) and cover-slipped.
MIN6 cells were seeded on attachment-factor-coated coverslips in a 12-well plate. Cells were washed, fixed with 4% PFA, permeabilised with 0.5% Triton and blocked in 2% BSA prior to incubating with primary antibodies overnight at 4°C (ESM Table 2). Cells were then incubated with fluorophore-tagged secondary antibodies for 1 h at room temperature, stained with DAPI and mounted onto microscope slides. Imaging was performed on an Olympus VS120 or Leica SP5 Confocal microscope (Olympus Life Science Solutions, Japan) and intensity or positive cell numbers were measured using ImageJ. Measurement of corrected total cell fluorescence is outlined in ESM Methods.
Western blotting
Cells were homogenised in cold RIPA buffer (Cell Signalling Technology) containing protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor cocktail (Roche Applied Science, Hercules, CA, USA). Protein was quantified using a DC assay (BioRad, Hercules, CA, USA), denatured using Laemmli SDS buffer heated to 95°C for 5 min, and subjected to SDS-PAGE and then transferred onto PVDF membrane (Millenium Science, VIC, Australia) as described previously [19]. Membranes were probed (antibodies are listed in ESM Table 2), visualised on an Odyssey CLx Imaging System (Licor, Lincoln, NE, USA) and band intensity evaluated using ImageJ normalised to the housekeeping gene.
RNA extraction and quantification by reverse-transcription quantitative PCR
For reverse-transcription quantitative PCR (RT-qPCR), RNA was extracted using ISOLATE-II RNA MiniKits (Bioline, London, UK) with on-column DNase treatment. RNA was quantified using a Nanodrop (BioTek, Winooski, VT, USA) and reverse-transcribed using a SensiFAST cDNA synthesis kit (Bioline). cDNA was amplified in triplicate with commercially available gene-specific hydrolysis probes (Taqman primers, listed in ESM Table 4, Invitrogen, Carlsbad, CA, USA) using a CFX384 PCR machine (BioRad). Thermal cycling conditions were 95°C for 2 min, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. Data were analysed using the \(^}_}}\) method, with expression normalised to the reference gene (18S) and also to the referent control group.
Single-cell RNA-seq analysis
Single-cell RNA-seq data were accessed from the Human Pancreas Analysis Program (HPAP-RRID:SCR_016202) Database (https://hpap.pmacs.upenn.edu/). Detailed methodology is outlined in ESM Methods.
Statistical analysis
Data are presented as the mean ± SD from at least three independent cell cultures, comprising n=3 or 4 animals or n=3–6 human samples per group. Differences between the two groups were assessed using two-tailed Student’s t tests. When comparing more than two groups, significance was calculated using one-way or two-way ANOVA, depending upon the number of independent variables, followed by Tukey’s post hoc testing for multiple comparisons, using GraphPad Prism software v.10 (San Diego, CA, USA). p<0.05 was considered statistically significant.

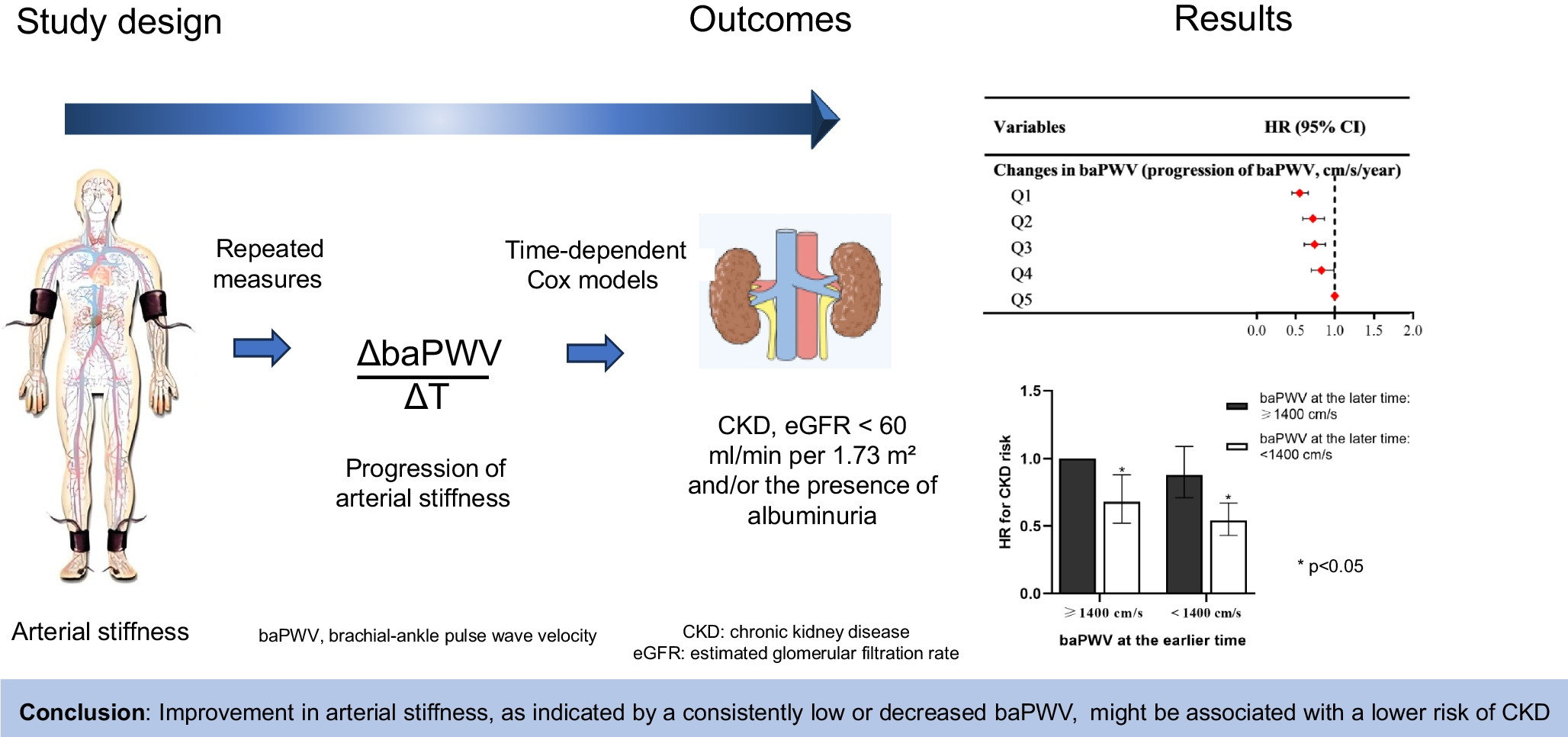

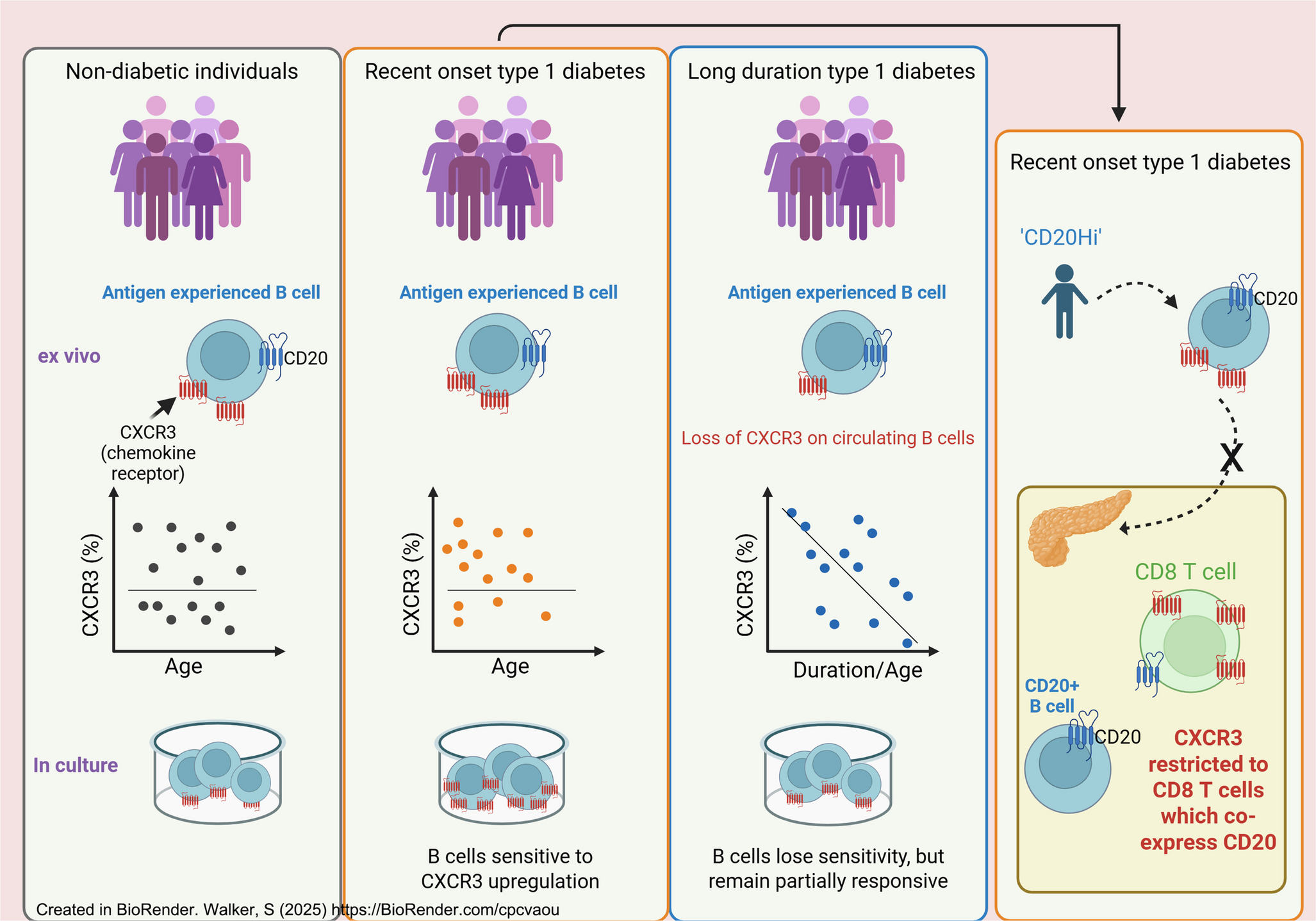
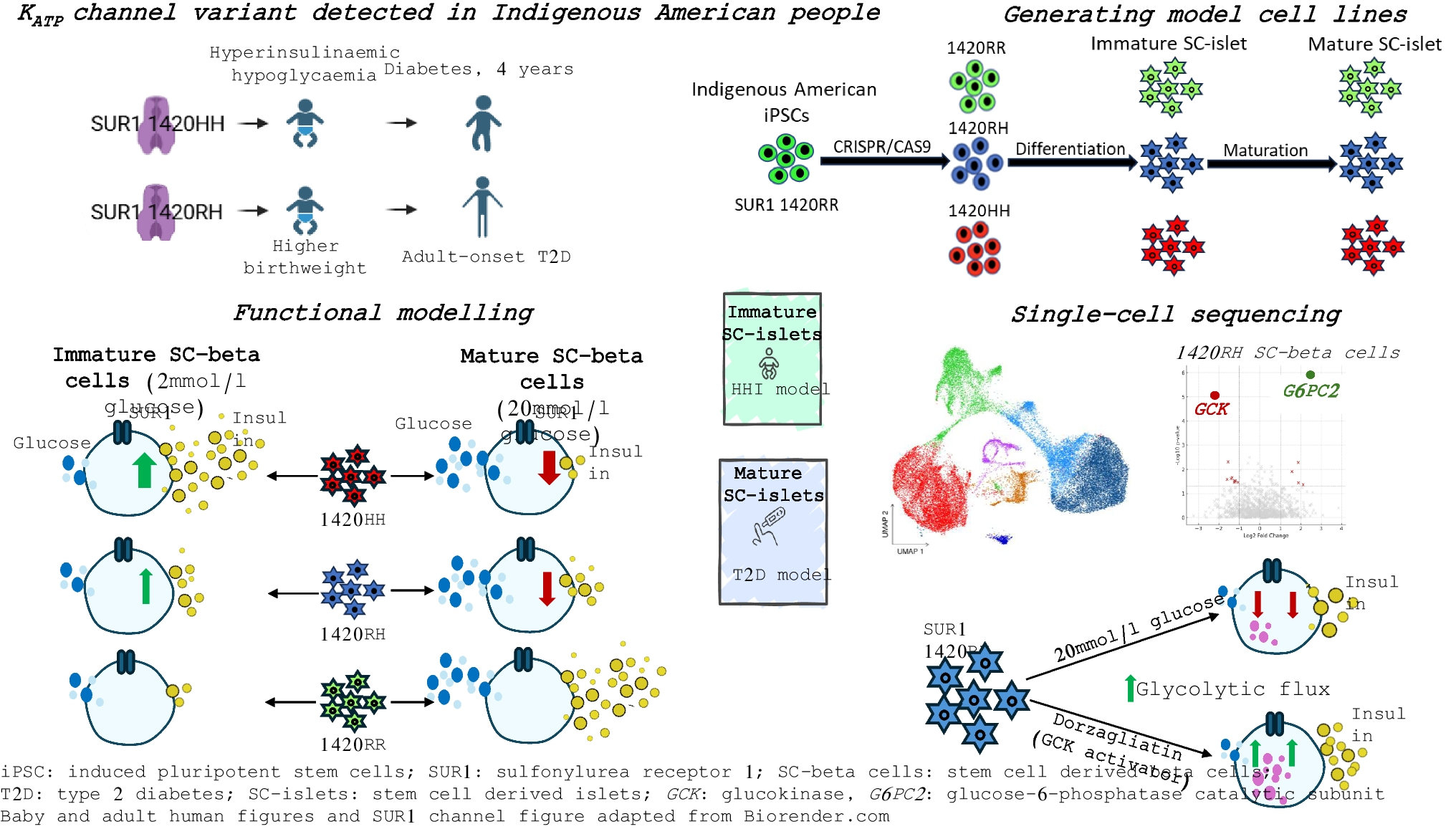

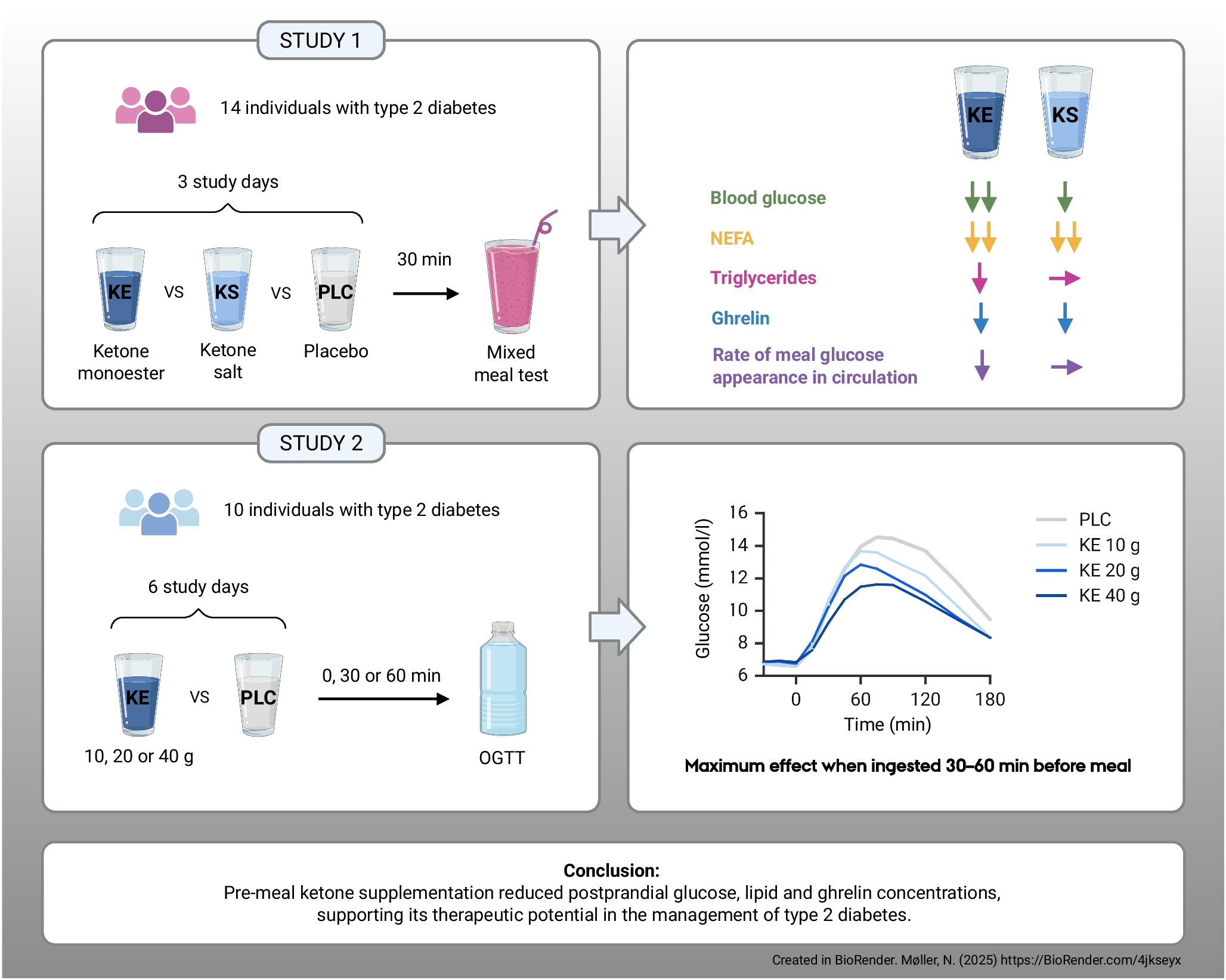
Comments (0)