
Remember me
This will be a pragmatic, participant-blind, parallel-group, randomised feasibility trial comparing access to a podiatry service with usual care. The trial is registered with the Australian and New Zealand Clinical Trial Registry (ANZCTRN12625001000493) and has been developed in accordance with the SPIRIT 2025 statement [17], the CONSORT 2010 statement extension to randomised pilot and feasibility trials [18], and the TIDieR checklist for reporting of interventions [19]. Ethics approval has been obtained from the La Trobe University Human Ethics Committee (Approval number HEC25095 on the 20th of May 2025) and written informed consent will be obtained from all participants during the baseline assessment.
The randomisation module in REDCap (Research Electronic Data Capture, Vanderbilt University, USA) will be used to randomise participants. Participants will be allocated on a 1 to 1 ratio with blocking in groups of 4 and 6, and stratification based on sex at birth to ensure a balance of participant sex between the groups. Allocation will be concealed from all researchers until after eligibility screening. Participants will be randomised to their intervention after completing consent at the baseline assessment.
Given this is a feasibility trial, only participants will be blind to their allocation. Both groups will be blind to group allocation through limited disclosure. All participants will be informed that two different types of treatment are being compared, however only the intervention group will be informed about the additional access to podiatry treatment.
Study groupsThe podiatry service will operate from the La Trobe University Health Sciences Clinic in Melbourne, Australia. Given this is a pragmatic trial, participant needs will be triaged during the baseline assessment, which will determine the types of interventions provided by the podiatry service. Based on previous research [20], it is anticipated that treatment will involve broad categories of:
1.dermatological care (nail and callus care, education);
2.musculoskeletal care (managing pain, deformity, and improve function using: (i) medication in conjunction with medical team, (ii) strengthening exercise and stretches, (iv) foot orthoses, (v) in-shoe padding, (vi) toe splints, and (vii) education);
3.neurovascular care (assessment, education, management where appropriate, and referral if needed);
4.foot ulcer management (pressure offloading, wound care and dressing, and referral where appropriate);
5.footwear education and modification.
Treatment will be provided by a podiatrist with over 10 years of experience in clinical practice, who is endorsed to prescribe scheduled medicines, has undertaken further training in rheumatological conditions, and plays an active role in the Australian Rheumatology Association. For participants who require foot orthoses, Formthotics™ (Foot Science International, Christchurch, New Zealand) full length prefabricated foot orthoses will be provided. These are manufactured from a dual-density polyethylene closed cell foam that has a firm-density bottom layer (Shore-A durometer 50), and a soft-density top layer (Shore-A durometer 25). Participants will be provided the foot orthoses in accordance with the manufacturer’s instructions by selecting an appropriate size, heating the foot orthoses using a specifically designed machine from Foot Science International, placing them within the participants footwear, and asking participants to stand to allow the foot orthoses to mould to the contour of the foot. Adjustments to the foot orthoses will be made to ensure the participants agree they are comfortable. Participants who require footwear will be provided with a $250 discount from the cost of new footwear. The footwear will be agreed with the podiatrist and participants will be required to attend pre-arranged footwear suppliers to receive the discount. Advice provided to participants about footwear is included in Online Resource 1. All other treatment, such as skin or nail care, exercises, or other aids, will be provided in line with standard podiatry practice in Australia.
The control group will receive usual care plus a self-management package based on advice from Creaky Joints Australia (29 Tips from Podiatrists for Managing Arthritis Pain in Your Feet), which provides a range of education including regular self-inspection of feet, advice about exercising to reduce foot/ankle discomfort, selecting appropriate footwear, and the use of supportive aids such as foot orthoses, ice packs, salt baths etc. For usual care, participants will be advised they can seek any treatment they prefer over the 12-week duration of the trial and their progress will be monitored. At the conclusion of the trial, participants randomised to the control group will be invited to attend a debriefing session and will be offered the same treatment as the intervention group if the intervention is found to be effective.
Patient and public involvementRecognising the importance of designing patient-centred research, we included a consumer as a coauthor (KL) during the initial design of the trial and securing grant funding. To further inform the trial design, we conducted four semi-structured interviews with people who have a rheumatological condition and foot-specific complaints. These interviews aimed to gather detailed feedback on various aspects of the trial, including the acceptability of the intervention, the feasibility of the proposed procedures, potential barriers to participation, and what outcomes are important. The interviews were conducted prior to the approval of the ethics application.
Participants will be recruited from outpatient rheumatology clinics at Austin Health in Melbourne, Australia. Rheumatologists will be informed about the trial and asked to recommend that their patients consider participating if they have foot-specific symptoms. Therefore, rheumatologists will initially approach participants about participating in the study and recommend that potential participants contact the researchers via a website. To be eligible for inclusion, participants must:
1.be adults over 18 years of age;
2.have a diagnosis of a rheumatological condition from a rheumatologist;
3.have foot-specific symptoms rated 3/10 or greater on a numerical rating scale (e.g. pain, stiffness, dermatological complaints); and
4.have experienced symptoms for at least four weeks.
Participants will be excluded if they:
1.are unable to understand the English language in verbal or written form;
2.have regularly attended a podiatry service for management of their rheumatological foot problem in the past 3 months;
3.have an active bacterial infection of the foot; and
4.experience foot symptoms that are not related to the rheumatological condition.
The recommended sample size for feasibility studies is 12 people per group [21], however to allow for a 20% drop-out rate, we will recruit 15 participants per group. This is similar to the median sample size of 30 for pilot trials documented in the United Kingdom Clinical Research Network database [22]. Given the low-risk nature of the interventions, we are not specifying interim analyses or guidelines for stopping the trial.
Main outcome variable: feasibilityGiven this is a pragmatic trial, the framework developed by Chan et al. [23] will be used to determine feasibility. According to this framework, 10 important domains of uncertainty should be addressed in a pragmatic feasibility trial. These domains are outlined in Table 1 along with criteria that will determine whether a larger trial is feasible. In addition to the feasibility outcomes, data will be collected on participant characteristics, adherence to interventions, adverse effects and acceptability and credibility of the interventions. These outcomes are outlined in detail in Online Resource 2.
Table 1 Domains and criteria used to evaluate trial feasibilityOther variables: limited efficacy testingIn addition to evaluating feasibility, limited efficacy testing will be undertaken on measures to be included in a fully powered randomised trial. These measures are outlined briefly in Table 2 and in specific detail in Online Resources 2.
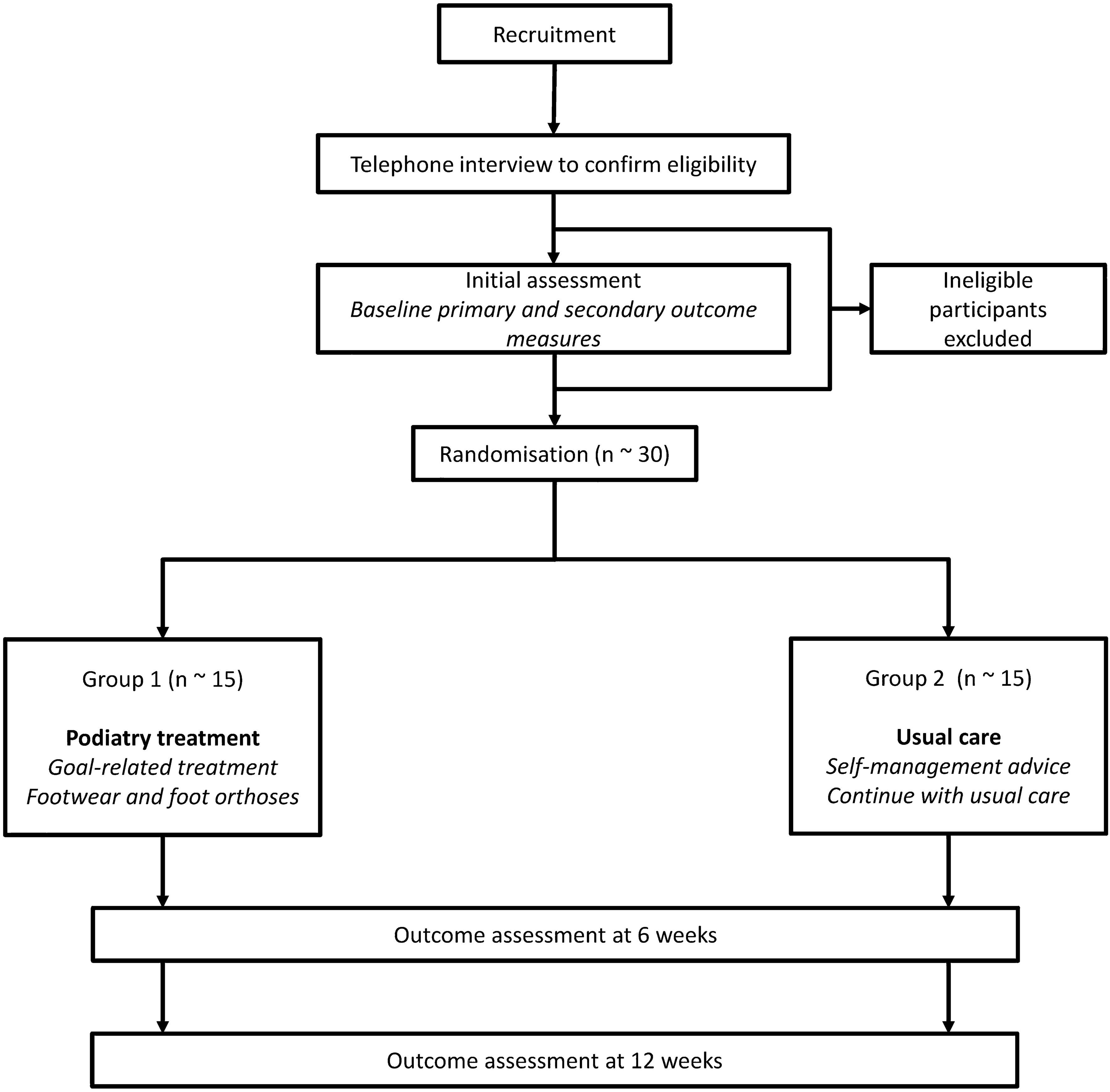
Table 2 Other variables measured for limited efficacy testingProceduresThe flow of participants through the trial is displayed in Fig. 1. Potential participants will be initially screened via telephone interview against the inclusion criteria by a research assistant, who will enter data into REDCap. The data collected during this screening telephone interview is outlined in Online Resource 3. During eligibility screening, if the participant is eligible for inclusion an appointment will be made for the baseline assessment with the Chief Investigator. At the baseline appointment, the Chief Investigator will randomise participants to either the intervention or control group after obtaining informed consent to participate in the trial.
Fig. 1
Diagram of participant flow
The total period of data collection for participants is 12 weeks. Participants who are enrolled in the trial will attend a baseline appointment at La Trobe University Health Sciences Clinic to complete outcome measures and commence podiatry treatment or receive the control intervention depending on their allocation. All outcomes will be completed on a computer and recorded in REDCap except for goals developed for Goal Attainment Scaling, which will be recorded on hard copy sheets for each individual participant. The change in Goal Attainment Scaling (rather than the goal itself) will be recorded by the Chief investigator in REDCap at the week 12 follow-up assessment. At week 6, the research assistant will send outcome measures to participants by email with a link to the REDCap survey. Follow-up assessment at week 12 will be in-person at the La Trobe University Health Sciences Clinic. Participants who are randomised to the intervention group will also attend treatment visits at the La Trobe University Health Sciences Clinic, which will occur depending on the treatment required.
Statistical analysisDescriptive statistics will be used to report feasibility outcomes. Mean (SD) scores and mean differences (95% CI) will be used to explore differences in continuous variables between the groups and Cohen’s d will be calculated to evaluate efficacy for the secondary outcome measures. A small effect size (Cohen’s d ≥ 0.20) will be regarded as an acceptable between group difference to signal efficacy. Data that is not continuous (adverse effects and use of cointerventions) will be reported using frequencies, medians and interquartile ranges as appropriate. Between group differences will be explored using risk ratios. Missing data will be minimised as much as possible, however no statistical approaches will be used to replace missing data. Any missing data will be evaluated and reported.
Comments (0)