
Remember me



The tumor microenvironment (TME) is no longer viewed solely as a collection of malignant and stromal cells but as a complex, multicellular ecosystem in which the nervous system plays a central regulatory role. Accumulating evidence demonstrates that both the peripheral and central nervous systems actively engage in bidirectional communication with tumor cells through a variety of molecular mechanisms. This neuro-tumoral crosstalk—mediated by neurotransmitters, neuropeptides, glial intermediaries, and structural remodeling—not only regulates cancer cell proliferation, invasion, and metastasis but also orchestrates immune evasion, metabolic reprogramming, and therapy resistance. Rather than being passive bystanders, neurons and their supporting glial cells function as dynamic architects of the TME, shaping tumor behavior at both local and systemic levels.
3.1 Direct paracrine and synaptic-like signalingOne of the most paradigm-shifting discoveries in cancer neuroscience is the formation of functional, synapse-like connections between neurons and tumor cells—termed neuro-gliomal synapses (NGS) in gliomas and neuron-to-cancer synapses in peripheral tumors. These structures enable rapid, activity-dependent signaling that mirrors classical neuronal communication.
In high-grade gliomas, glutamatergic neurons form excitatory synapses with tumor cells via AMPA-type glutamate receptors (AMPARs), particularly those containing GluA1/GluA2 subunits. Upon synaptic activation, AMPARs mediate Na⁺ influx and membrane depolarization in glioma cells, triggering calcium entry through voltage-gated calcium channels (VGCCs). This Ca²⁺ surge activates downstream effectors such as calcium/calmodulin-dependent kinase II (CaMKII) and transcription factors including CREB, MYC, and FOS, driving tumor proliferation and network integration [28]. Electrophysiological recordings and optogenetic stimulation have confirmed the functionality of these synapses, demonstrating that neuronal activity directly fuels oncogenic programs in real time.
Beyond glutamate, other neurotransmitters exert potent effects via receptor-mediated signaling. Norepinephrine (NE), released by sympathetic nerves, binds β-adrenergic receptors (β-ARs)—predominantly β₂-AR—on tumor cells in breast, ovarian, and prostate cancers. This activates Gs-protein-coupled cAMP/PKA/CREB signaling, promoting survival (via upregulation of BCL2 and MCL1), epithelial-mesenchymal transition (EMT), and migration [3, 29]. Parallel activation of PI3K/Akt/mTOR and MAPK/ERK pathways further enhances tumor growth and invasive potential.
Parasympathetic and sensory inputs add layers of complexity. Acetylcholine (ACh) from vagal or local cholinergic nerves binds muscarinic receptors (e.g., CHRM1) on gastric and pancreatic cancer cells, activating Gq-coupled signaling that increases intracellular Ca²⁺ and PKC activity, ultimately stimulating PI3K/Akt/mTOR to promote proliferation [30]. Conversely, in colorectal cancer (CRC), ACh acting on α7nAChR-expressing macrophages initiates the “cholinergic anti-inflammatory pathway,” suppressing NF-κB activation and pro-inflammatory cytokine production (TNF-α, IL-6), thereby reducing inflammation-driven tumorigenesis [20].
Sensory neurons release neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P (SP). CGRP binding to CLR/RAMP1 complexes on tumor cells activates cAMP/PKA and ERK pathways, promoting proliferation and immune evasion [31]. SP, via neurokinin-1 receptor (NK1R), enhances motility and angiogenesis by inducing VEGF expression in tumor and endothelial cells [32].
Critically, these signaling axes are not isolated but often converge and cross-regulate, creating a dense molecular network that integrates neural activity with oncogenic signaling.
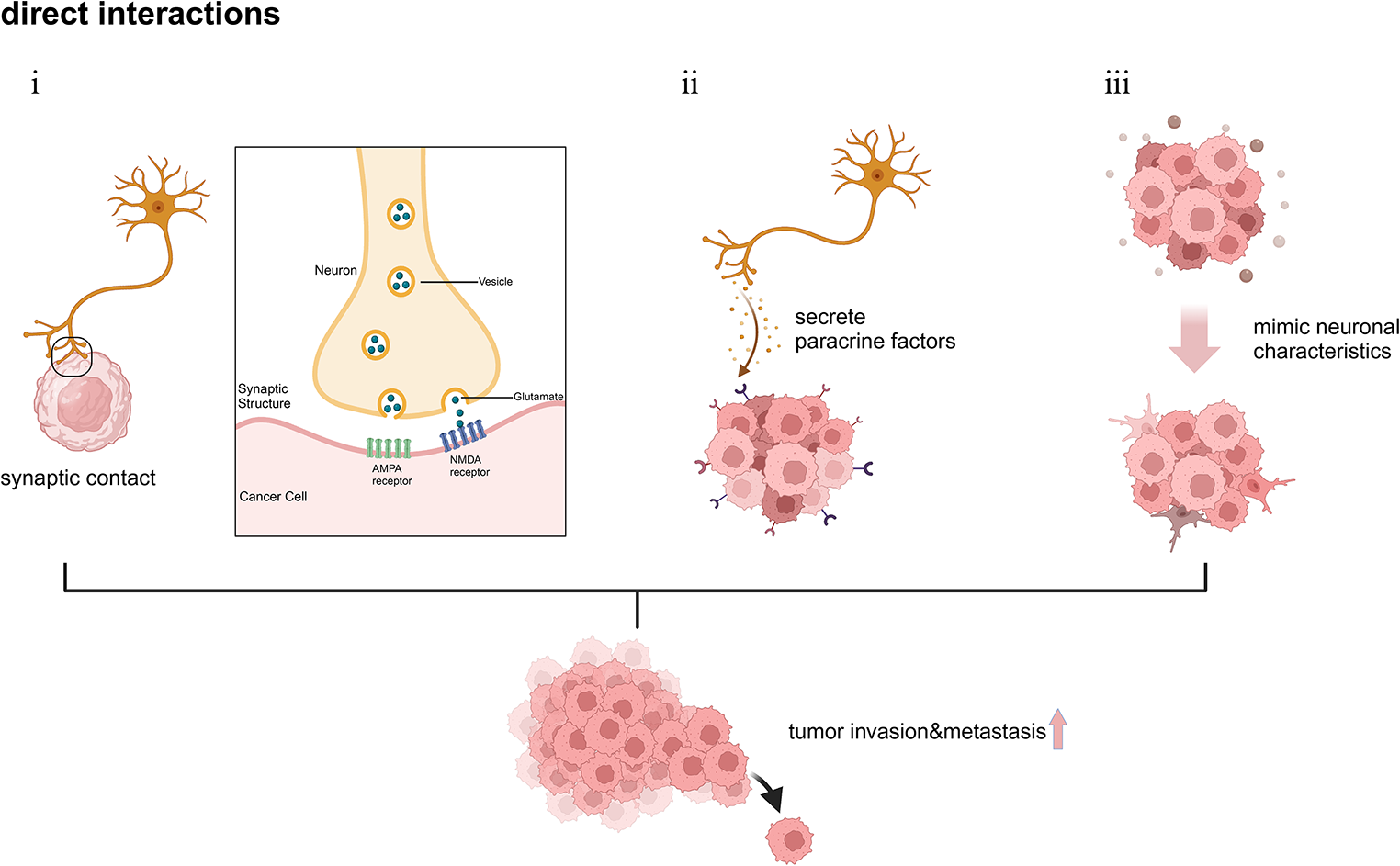
Fig. 1
Schematic illustration of the direct interactions between tumor cells and neurons. (i) Neurons interact with tumor cells through synaptic contact, activating postsynaptic receptors such as AMPA receptors and NMDA receptors. (ii) Neurons promote tumor metastasis and invasion through paracrine signaling pathways. (iii) Tumor cells mimic neurons to acquire enhanced invasiveness and facilitate metastasis
3.2 The pivotal role of Schwann cells and cancer-associated fibroblasts (CAFs)Schwann cells (SCs) and cancer-associated fibroblasts (CAFs) have emerged as central intermediaries in neuro-tumoral crosstalk, forming a critical “bridge” that connects nerves to cancer cells.
SCs are reprogrammed by tumor-derived signals. Single-cell and spatial transcriptomic analyses have identified distinct, pro-invasive SC subtypes, such as TGFBI SCs localized at the forefront of perineural invasion (PNI), which can be induced by TGF-β and promote tumor cell migration [33] A key mechanism activating both SCs and tumor cells is integrin signaling. Vitamin D binding protein(GC) acts as a novel ligand for integrinβ1(ITGB1), activating both tumor cells and SCs via the ITGB1-FAK axis to promote PNI in PDAC 31 This signaling enhances SC-mediated ECM remodeling and creates a chemotactic gradient, guiding tumor cells along nerve tracts [34]. SCs guide PNI through multiple mechanisms: (i) secreting neurotrophic factors (e.g. NGF, BDNF, GDNF) to create chemotactic gradients that attract tumor cells expressing cognate receptors (TrkA, RET) [33, 35]; (ii) upregulating cell adhesion molecules (e.g. L1CAM, N-cadherin) to stabilize heterotypic interactions with tumor cells [35]; and (iii) remodeling the extracellular matrix (ECM) by depositing laminins and secreting matrix metalloproteinases (MMPs) to generate permissive “highways” for dissemination [33, 34]. Integrin signaling is a key activator; vitamin D binding protein (GC) acts as a ligand for integrin β1 (ITGB1), activating both tumor cells and SCs via the ITGB1-FAK axis to promote PNI in PDAC [34]. Beyond the well-established neurotrophic factor gradients, cancer cell-intrinsic programs also critically govern PNI. For instance, in colorectal cancer, the cancer-testis antigen MAGEA6 engages the transcription factor YY1 to orchestrate a transcriptional program that dictates perineural invasion, highlighting a novel cell-autonomous mechanism driving this process [9]. Furthermore, non-myelinating SCs (NMSC) are multifaceted regulators of the TME in gastrointestinal cancers, their enrichment correlating with core cancer hallmarks like “Activating Invasion and Metastasis [36].
CAFs actively participate and form positive feedback loops. In colorectal cancer (CRC), norepinephrine (NE) activates ADRB2 on CAFs, inducing their secretion of nerve growth factor (NGF). Increased NGF further promotes intratumoral sympathetic innervation and NE accumulation, thereby accelerating CRC growth via ADRA2A/Gi-mediated YAP signaling activation, forming a coherent β2-adrenergic-NGF feedforward loop [37]. Beyond soluble factors, CAF-derived extracellular vesicles (EVs) can package RNAs such as the PNI-associated transcript (PIAT). PIAT binds to YBX1 and stabilizes pro-PNI mRNAs in an m5C modification-dependent manner, driving neural remodeling [38]. CAFs also foster a high-lactate microenvironment [39], among other mechanisms, to multidimensionally support PNI and tumor progression.
3.3 Glial and Schwann cell-mediated interactionsNon-neuronal glial cells serve as essential intermediaries in neuro-tumoral crosstalk, facilitating physical invasion, trophic support, and microenvironmental remodeling.
In peripheral cancers such as pancreatic, prostate, and head and neck carcinomas, Schwann cells (SCs) play a pivotal role in enabling perineural invasion (PNI)—a pathological hallmark associated with poor prognosis. Upon exposure to tumor-derived signals, SCs undergo phenotypic reprogramming into a repair-like state reminiscent of “Bands of Büngner” during nerve regeneration. These activated SCs secrete neurotrophic factors—including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glial cell line-derived neurotrophic factor (GDNF)—creating chemotactic gradients that attract tumor cells expressing TrkA, TrkB, or RET receptors [40].
Additionally, SCs upregulate cell adhesion molecules such as L1CAM and N-cadherin, which bind corresponding ligands on tumor cells, stabilizing heterotypic interactions and guiding directional migration along nerve tracts. They also remodel the extracellular matrix (ECM) by depositing laminins and secreting matrix metalloproteinases (MMPs), generating permissive “highways” for tumor dissemination.
Single-cell RNA sequencing has identified distinct subpopulations of tumor-associated SCs with pro-invasive transcriptomic signatures enriched in axon guidance, ECM organization, and cytokine signaling pathways. Notably, interleukin-33 (IL-33), released from injured nerves, activates SCs to produce CCL2, which recruits tumor-associated macrophages (TAMs), forming a tripartite IL-33/SC/TAM axis that exacerbates PNI and fosters local immunosuppression [40].
In the central nervous system, astrocytes adopt dual roles depending on context. Reactive astrocytes surrounding gliomas form a supportive scaffold that enhances tumor invasion by secreting IL-6, TGF-β, and TIMP-1, which promote EMT-like transitions, stemness, and apoptosis resistance [41]. Gap junction-mediated transfer of metabolites (e.g., lactate, glutathione) and second messengers (e.g., cGAMP) between astrocytes and glioma cells further contributes to therapy resistance and metastatic spread [42, 43].
However, under certain conditions—such as low-grade lesions or specific immune contexts—astrocytes can exert tumor-suppressive effects by releasing exosomal miRNAs that silence oncogenes or by activating anti-tumor immune responses. This duality underscores the context-dependent nature of glial influence in cancer progression.
3.4 Neuro-immune crosstalk in the TMENeural inputs profoundly reshape the immune landscape of the TME, often tipping the balance toward immunosuppression.
Sympathetic activation via β-AR signaling on myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) induces an M2-like, immunosuppressive phenotype characterized by elevated expression of IL-10, ARG1, and PD-L1 [44, 45]. This impairs antigen presentation and dampens CD8⁺ T-cell responses. Chronic stress amplifies this effect, correlating with reduced T-cell infiltration and accelerated tumor growth—effects reversible with β-blockers like propranolol [46].
Sensory neurons also modulate dendritic cell (DC) function in a context-dependent manner. For instance, in melanoma, CGRP released from peptidergic nociceptors can contribute to an immunosuppressive microenvironment by directly promoting the exhaustion of cytotoxic CD8 T cells [25, 47]. Furthermore, signaling through the CGRP receptor RAMP1 on intratumoral CD8 T cells is associated with increased exhaustion and poorer clinical prognosis. Genetic ablation of nociceptors or antagonism of RAMP1 reduces T cell exhaustion and decreases tumor growth. Conversely, in the gastrointestinal tract, CGRP can paradoxically promote anti-tumor immunity through distinct mechanisms.
Paradoxically, some neural circuits enhance anti-tumor immunity. Dorsey et al. showed that TRPV1⁺ sensory neurons in the gut sense microbial signals and release CGRP, which induces CCL2 secretion from stromal cells, recruiting IL-17-producing γδ T cells. These cells secrete IFN-γ and exert potent anti-tumor activity in early-stage colorectal cancer—a rare example of neural-enhanced anti-tumor immunity.
Furthermore, the vagus nerve-mediated cholinergic anti-inflammatory pathway, acting through α7nAChR⁺ macrophages, suppresses NF-κB activation and TNF-α production, protecting against colitis-associated CRC [48]. Electrical vagus nerve stimulation (VNS) has been shown to reduce tumor-promoting inflammation in preclinical models, highlighting the therapeutic potential of harnessing this pathway [49], It is crucial to note, however, that the role of vagal signaling in cancer is not universally protective and exhibits profound context- and tumor type-specific effects.
Together, these findings illustrate that the nervous system functions as a master regulator of tumor immunity, capable of either suppressing or enhancing immune responses based on neural subtype, neurotransmitter profile, and tissue context. The neuro-immune cross-talk within the TME is multifaceted, involving direct neuronal interactions with immune cells and indirect modulation via cytokine and chemokine networks [50].
The nervous system’s regulation of natural killer (NK) cells is a recently uncovered and crucial mechanism. In pancreatic ductal adenocarcinoma (PDAC), nociceptor neurons interact with cancer-associated fibroblasts (CAFs) to suppress CAF-derived IL-15 secretion, thereby impairing NK cell tumor infiltration and cytotoxic function, which drives PDAC progression and cancer pain [11].
More profoundly, cancer-induced nerve injury (CINI) itself has been identified as a novel mechanism of resistance to immunotherapy. Cancer cell-mediated damage to nerve myelin sheaths triggers neurons to autonomously initiate an IL-6 and type I interferon-mediated inflammatory response. This leads to a globally immunosuppressive and exhausted tone within the TME, thereby promoting resistance to anti-PD-1 therapy [51]. This work mechanistically linked neuronal damage to a broad, TME-wide state of immune dysfunction, providing a strong rationale for combining neuroprotective strategies with immunotherapy. This provides a strong rationale for combining neuromodulatory strategies with immune checkpoint blockade.
Intriguingly, neurodegeneration of local sympathetic inputs may paradoxically promote cancer progression in some contexts. In colorectal cancer (CRC), norepinephrine (NE) acts via α2-adrenergic receptors (ADRA2) on tumor cells to recruit CD8 T cells. The degeneration of sympathetic nerves reduces this form of anti-tumor immune surveillance [52], highlighting the complexity and context-dependence of neuro-immune regulation.
3.5 Structural and metabolic remodeling by neural inputsBeyond biochemical signaling, neuronal activity drives large-scale architectural and metabolic changes in the TME.
Axonal fibers provide physical highways for tumor cell migration during perineural invasion (PNI). Tumor cells align along nerve bundles, using them as low-resistance routes for deep tissue invasion—a process facilitated by Schwann cell-mediated ECM remodeling and neurotrophin gradients. In colorectal cancer (CRC), perineural invasion (PNI) is increasingly recognized as a clinically significant event, associated with higher rates of local recurrence, distant metastasis, and reduced survival. The molecular underpinnings of CRC-PNI involve reactivation of developmental axon guidance pathways, such as Netrin-1/DCC and Slit/Robo, which promote tumor neurotropism and directional migration along nerves. Additionally, neurotrophic factors (e.g., NGF, GDNF) secreted by tumor and stromal cells drive neurite sprouting, while tumor-expressed adhesion molecules like L1CAM facilitate direct interactions with Schwann cells and axons. These coordinated mechanisms enable cancer cells to exploit nerves as conduits for dissemination, reinforcing the role of the nervous system as an active participant in structural remodeling of the TME [53].This spatial guidance mimics developmental axon pathfinding and involves several key molecular axes:
(1) NGF/TrkA/p75NTR: Nerve growth factor (NGF), secreted by tumor cells and reactive stromal cells, acts as a potent chemoattractant for sensory and sympathetic axons. It binds to its high-affinity receptor TrkA and low-affinity receptor p75NTR on neurons, promoting axonal sprouting toward tumors—a phenomenon termed neoneurogenesis. Notably, this process is bidirectional: in turn, newly formed nerves release neurotransmitters such as norepinephrine and neuropeptide Y that enhance tumor cell invasiveness, stemness, and perineural niche formation.
(2) CXCL12/CXCR4 & Netrin-1/DCC: The chemokine CXCL12, produced by perineural fibroblasts and Schwann cells, establishes a concentration gradient that guides CXCR4-expressing tumor cells along nerves. Similarly, netrin-1, secreted by neurons and tumor cells, interacts with its receptor DCC (deleted in colorectal cancer) to mediate axon-like navigation of cancer cells, promoting directional migration and neurotropic invasion. These ligand-receptor pairs function as “molecular compasses” that coordinate reciprocal neuron-cancer trafficking.
(3) Integrin/FAK/YAP: Tumor cells adhere to laminin-rich extracellular matrix (ECM) surrounding nerves via integrins (e.g. α6β1, α6β4), which cluster at focal adhesions and activate focal adhesion kinase (FAK). FAK signaling triggers cytoskeletal reorganization and activates downstream effectors such as Src and YAP, driving invasive motility and resistance to anoikis. The physical properties of the tumor microenvironment, influenced by neural activity, also impact PNI. A stiffened ECM signals through the integrin β1 (ITGB1)-FAK-YAP axis to induce the expression of pro-metastatic and neurotrophic genes (e.g., NGF) in breast cancer cells, ultimately enhancing perineural invasion [27], This integrin-mediated anchoring allows cancer cells to maintain traction during migration along nerve tracts and contributes to the establishment of a pro-invasive perineural niche.
At the systemic level, central autonomic nuclei such as the paraventricular nucleus (PVN) of the hypothalamus and the central amygdala (CeA) integrate emotional and physiological stimuli (e.g., stress, sleep disruption) and relay signals via sympathetic outputs to influence tumor metabolism. β-AR signaling reprograms cancer cell metabolism toward glycolysis and oxidative phosphorylation, increasing bioenergetic fitness [4], In prostate cancer, adrenergic signaling induces an angio-metabolic switch that supports tumor growth [21].
Neural inputs also modulate vascular permeability and lymphangiogenesis. NE and SP increase VEGF expression in tumor and stromal cells, promoting angiogenesis and potentially compromising the blood-brain barrier in brain tumors—offering both challenges and opportunities for drug delivery.
Emerging data even suggest that bioelectric fields generated by neuronal firing may influence tumor cell polarity and collective migration, adding a biophysical dimension to neural regulation. Glioblastoma (GBM) cells modulate neuronal excitability through ion channels. By expressing the potassium channel Kv4.2, GBM cells alter the extracellular potassium concentration, enhancing the excitability of surrounding neurons. This directly links tumor-associated epilepsy to tumor progression [54].
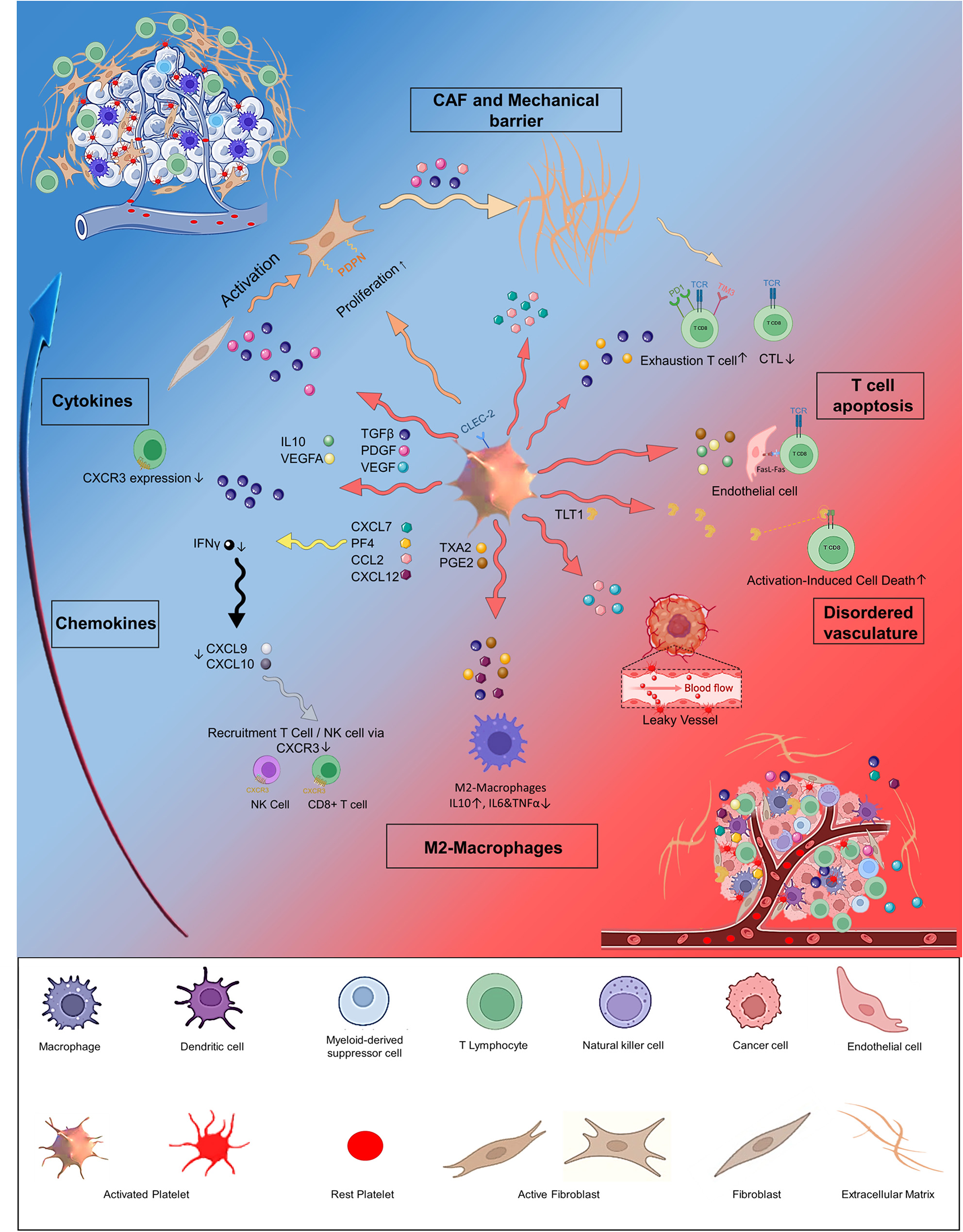
Fig. 2
Schematic illustration of the interactions between TME and neurons. Neurons indirectly promote tumor progression by interacting with components of the tumor microenvironment (TME), such as the extracellular matrix, lymphatic vessels, blood vessels, and immune cells
The intricate mechanisms outlined above—ranging from direct synaptic communication to complex neuro-immune-stromal interactions—collectively unveil a multitude of therapeutic vulnerabilities. We now explore the translational opportunities arising from this knowledge.
Comments (0)