2.1 Patients and samples
A total of 272 patients diagnosed with colorectal cancer and underwent surgical resection at the First Affiliated Hospital of Sun Yat-sen University (FAH-SYSU) between 2014 and 2019 were retrospectively included in this study. Primary colorectal tumor tissues, adjacent normal tissues and liver metastases were collected intraoperatively and immediately stored at –80 °C. Overall survival (OS) was calculated from the surgical date until mortality from any cause or the final follow-up visit. Progression-free survival (PFS) was measured from the operation date to the first documented disease progression or death from any cause, whichever came first. Patients without observed progression or death were censored at the date of their most recent follow-up. This study was approved by the Institutional Ethics Committee of FAH-SYSU (Approval number: [2022]378), and all participants provided written informed consent.
2.2 Cell lines and cell culture
The human colorectal cancer cell lines DLD-1 and HCT116 were obtained from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (PS, Gibco). All cells were maintained in a humidified incubator (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C with 5% CO2.
Stable METTL1-knockout CRC cell lines were generated using the lentiCRISPR v2 system. Cells were transduced with lentiCRISPR v2-sgControl (non-targeting, sgNC) as a single-guide RNA (sgRNA) negative control or lentiCRISPR v2-sgRNAs targeting METTL1 (sgMETTL1), followed by puromycin selection to establish stable cell lines. Knockout efficiency was confirmed by western blot analysis.
2.3 Animal experiments
Male NCG mice aged 5–6 weeks were obtained from GemPharmatech (Jiangsu, China). All animals were maintained under specific pathogen-free (SPF) conditions with standard husbandry. All the animal experiments were approved by the Animal Care and Use Committee of FAH-SYSU and complied with institutional and national ethical guidelines. Mice were routinely monitored and humanely euthanized upon reaching predefined endpoints based on tumor burden or overall health condition.
To establish subcutaneous xenografts, 1 × 10⁷ CRC cells transduced with control or METTL1-targeting sgRNA were suspended in 100 µL PBS and injected into the right flank of each mouse. Tumor size and body weight were monitored at regular intervals. Tumor volume was determined using the formula: length × width²/2. At the experimental endpoint, mice were euthanized, and tumors were harvested, weighed, fixed in formalin, or flash-frozen in liquid nitrogen for subsequent analysis.
For the intrasplenic injection liver metastasis model, 1 × 10⁶ CRC cells suspended in 100 µL of PBS were injected into male NCG mice. Three weeks post-injection, the mice were euthanized, and liver tissues were collected. Hematoxylin and eosin (H&E) staining was used to assess metastatic burden, while IHC was performed to assess METTL1 and Ki67 expression levels in liver metastases.
2.4 Proteomic sequencing and analysis
Proteins were precipitated, digested with trypsin, reduced, and alkylated, followed by LC-MS/MS on an EASY-nLC 1200 system coupled to an Orbitrap Exploris 480 mass spectrometer with FAIMS in data-dependent acquisition mode. Raw data were processed with Proteome Discoverer (v2.4.1.15), and proteins were identified and quantified against the human UniProt database with a false discovery rate (FDR) < 1%.
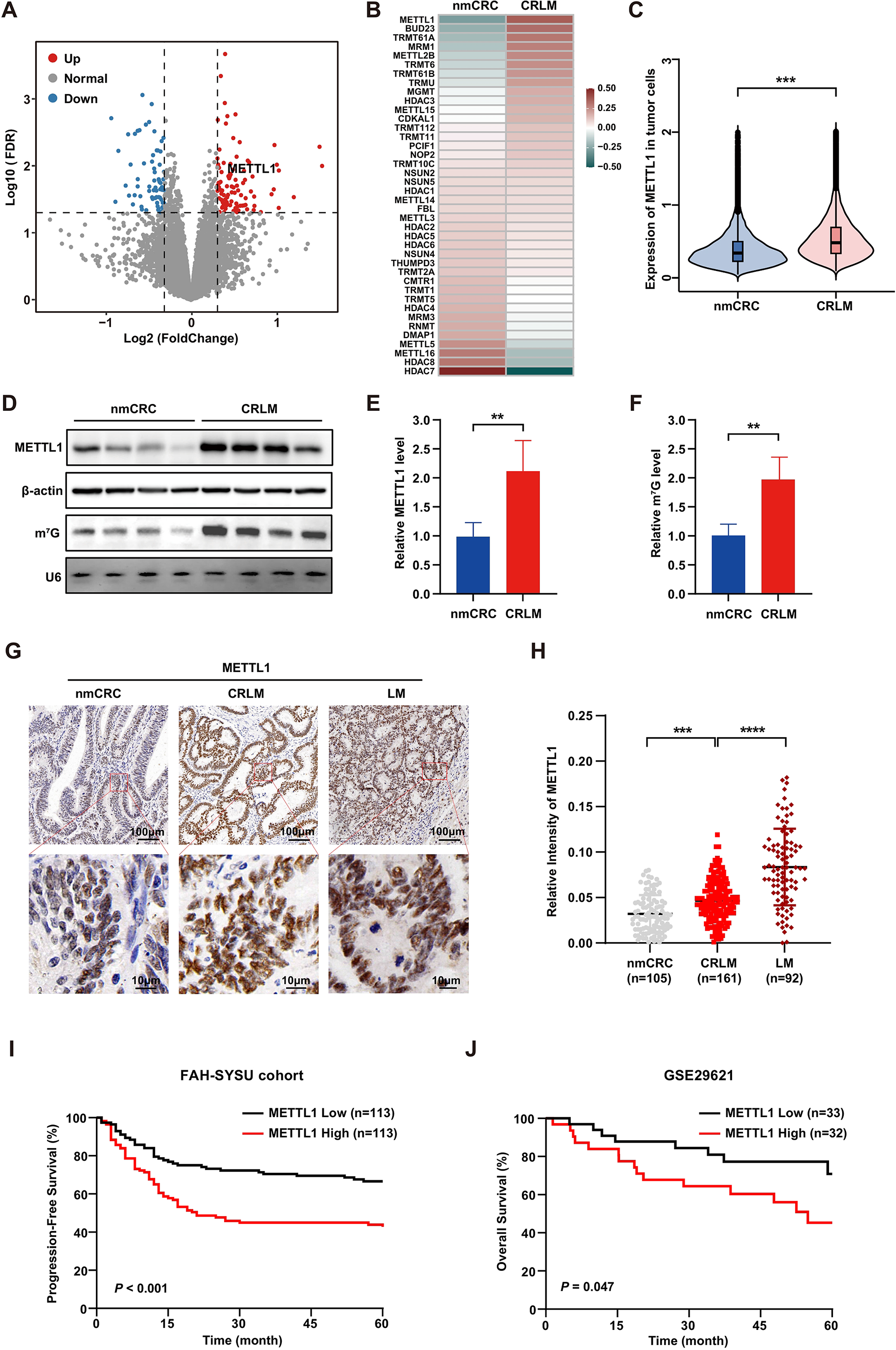
Proteomic data were normalized by median centering across all proteins. Proteins with more than 50% missing values were excluded prior to imputation. Missing values were imputed using the k-nearest neighbor (k-NN) algorithm. The limma R package was used to identify differentially expressed proteins between METTL1-knockout and control CRC cells. Genes were ranked in descending order according to their log2 fold change between the two groups to generate a gene list, which was subsequently subjected to gene set enrichment analysis (GSEA) and visualized using the clusterProfiler R package. Gene sets for GSEA were obtained from the Molecular Signatures Database (MSigDB).
2.5 Polysome profiling
Polysome profiling was conducted following established methodologies [11, 17]. Briefly, cells were collected and lysed on ice for 10 min in 1.2 mL of polysome lysis buffer composed of 150 mM NaCl, 50 mM MOPS (pH 7.4), 15 mM MgCl₂, 100 µg/mL cycloheximide, 1 mg/mL heparin, 0.5% Triton X-100, 2 mM PMSF, 200 U/mL RNase inhibitor, and 1 mM benzamidine. Lysates were centrifuged at 13,000 × g at 4 °C for 10 min to remove insoluble material. Subsequently, 1 mL of the supernatant was layered onto an 11 mL sucrose gradient (10%–50%) prepared in polysome buffer (50 mM MOPS, 15 mM MgCl2, 150 mM NaCl, and 100 µg/mL cycloheximide). The gradients were ultracentrifuged at 36,000 rpm for 3 h at 4 °C using the SW41 rotor in a Beckman Coulter ultracentrifuge (Beckman Coulter, USA). Gradients were fractionated using a BR-188 Density Gradient Fractionation System (Brandel, USA) with continuous monitoring of absorbance at 254 nm to resolve ribosomal subunits and polysome profiles.
2.6 Puromycin intake assay
The puromycin intake assay was carried out as reported previously [11, 18]. Cells were transfected with METTL1-specific small interfering RNA (siMETTL1) or non-targeting control siRNA (siNC) for 48 h. Subsequently, cells were treated with 1 µM puromycin for 30 min to enable incorporation into nascent polypeptide chains. Then cell lysis was performed and the protein lysates were subjected to western blot analysis using an anti-puromycin antibody to evaluate global translational activity.
2.7 Northern, northwestern, and western blot assays
Northern, northwestern, and western blot assays were conducted using previously described methods [11, 17]. For northern blotting, 2 µg of total RNA was resolved on a 10% TBE-UREA denaturing polyacrylamide gel (Bio-Rad, Hercules, CA, USA) to separate small RNAs such as tRNAs and U6 snRNA. Then the RNAs were transferred to positively charged nylon membranes (e.g., Hybond-N+, GE Healthcare) via electroblotting or capillary transfer, UV-crosslinked for 3 min, and hybridized with a digoxigenin (DIG)-labeled probe specific for U6 snRNA, according to the manufacturer’s instructions. For northwestern blotting, membranes were blocked and incubated overnight at 4 °C with an anti-m7G antibody (MBL International, Woburn, MA, USA). Signal detection for DIG-labeled probes and antibody-bound signals was performed using chemiluminescent substrates following standard protocols for western blot detection.
2.8 RNA isolation, reverse transcription and quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 2 µg of total RNA using the PrimeScript RT Reagent Kit (Takara, Japan) in a 20 µL reaction system, and diluted 1:20 for qRT-PCR. qRT-PCR was performed with TB Green Premix Ex Taq II (Takara, Japan) on a LightCycler 480 system (Roche, USA). Each sample was analyzed in triplicate, and relative mRNA levels were normalized to GAPDH.
2.9 m7G tRNA reduction and cleavage sequencing (TRAC-seq)
TRAC-seq was performed following previously reported procedures [11, 17]. Total RNA was extracted from CRC cells with or without METTL1 knockout, and then small RNAs were enriched using the mirVana™ miRNA Isolation Kit (Invitrogen, USA). These small RNAs were subjected to demethylation treatment with either recombinant wild-type or D135S mutant AlkB proteins. Half of the demethylated small RNAs were reserved as input control for small RNA library construction to assess tRNA expression levels. The remaining AlkB-treated RNAs were treated with 0.1 M NaBH₄ on ice in the dark for 30 min, using 1 mM free m7GTP as the methylation carrier. RNAs were then reacted in 100 µL of cleavage solution (H2O: glacial acetic acid: aniline = 7:3:1) for 2 h at room temperature in darkness to induce site-specific cleavage at m7G-modified sites. RNA purification was performed with the Oligo Clean & Concentrator kit (ZYMO Research, USA). Both input and aniline-cleaved RNAs were used to generate cDNA library with the Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs, USA), followed by high-throughput sequencing on the Illumina NextSeq 500 platform.
2.10 Ribosome-nascent chain complex-bound mRNA sequencing (RNC-seq)
RNC-seq was conducted based on established methodologies [11, 18]. Cells were treated with ice-cold PBS supplemented with 100 µg/mL cycloheximide to arrest ribosomes on mRNAs. Cells were then lysed on ice for 30 min in 1.2 mL of lysis buffer containing 1% Triton X-100 in ribosome buffer (RB buffer), composed of 15 mM MgCl₂, 20 mM HEPES-KOH (pH 7.4), 2 mM dithiothreitol (DTT), 200 mM KCl, and 100 µg/mL cycloheximide. Lysates were clarified by centrifugation at 16,200 × g for 10 min at 4 °C. 10% of the cleared supernatant was retained as input control. The remaining supernatant was layered onto 35 mL of sucrose cushion (30% sucrose in the RB buffer) and ultracentrifuged at 174,900 × g for 5 h at 4 °C using the SW32 rotor (Beckman Coulter, USA), enabling the isolation of ribosome-nascent chain complex (RNC) pellets enriched in polysomes. Total RNA from both input and RNC fractions was extracted as previously described. RNC-seq libraries were prepared and sequenced on the BGISEQ-500 platform by Beijing Genomics Institute (BGI, Shenzhen, China).
2.11 RNC-seq data analysis
RNA-seq data were processed according to established protocols [11, 18]. Sequencing reads were aligned to the human reference genome (GRCh38) using Bowtie2, and gene expression quantification was performed based on FPKM (Fragments Per Kilobase of transcript per Million mapped reads) method. Translation efficiency (TE) was calculated for each gene as the ratio of FPKM values from RNC-seq to those from input RNA-seq. Genes with significantly reduced TE were subjected to KEGG pathway enrichment analysis using the R package clusterProfiler.
2.12 Statistical analysis
Statistical analysis was performed using GraphPad Prism V9.5 and R version 4.4.3. Data are presented as mean ± standard deviation (SD) unless otherwise indicated. Statistical significance is represented as ns (not significant), *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Unless otherwise noted, comparisons of quantitative variables between two groups were performed using unpaired, two-tailed Student’s t-tests or Mann-Whitney U tests. Comparisons of categorical variables were performed using the chi-square test or Fisher’s exact test. For multiple group comparisons, one-way or two-way ANOVA followed by post hoc tests was performed as appropriate. Survival curves were generated using the Kaplan–Meier method and compared using the log-rank test.
Further methodological details and a list of reagents and resources used in this study are provided in Supplementary Material 1 (Supplementary Methods, Supplementary Tables 1 and 2).




Comments (0)