2.1 Data source
All datasets analyzed in this study are publicly accessible from reputable repositories. Raw single cell sequencing data was downloaded from the Genome Sequence Archive at the National Genomics Data Center (Beijing, China) under the BioProject ID PRJCA007744 [20]. Another processed scRNA-seq dataset, GSE149614, were downloaded from GEO data portal. Bulk RNA-seq datasets were retrieved from the TCGA-LIHC cohort via the UCSC Xena portal, the LIRI cohort from the ICGC data portal, the CHCC cohort from the NODE database, E-TABM-36 from EMBL’s European Bioinformatics Institute [21, 22], and GSE14520 [23], GSE76427 [24], GSE84005 [25] and GSE54236 [26] from the GEO database. Collectively, these datasets provide a comprehensive resource for investigating transcriptomic alterations associated with HCC (Supplementary Table S1).
2.2 scRNA-seq data pre-processing and quality control
Single-cell RNA-seq data were aligned and quantified using the CellRanger toolkit (v3.1) against the GRCh38 reference genomes. Low-quality empty droplets were removed using the emptyDrops function of the dropletUtils R package (v1.10.3), which assesses whether the RNA content associated with a barcode is significantly different from the ambient background RNA in each library. Cells with a Benjamini-Hochberg-corrected false discovery rate (FDR) < 0.01 were retained for further analysis. Cells were retained if they contained fewer than 20% mitochondrial gene content, expressed more than 200 genes, and had between 200 and 6000 detected genes, appearing in at least three cells. After quality control, 360,046 cells were retained [20].
2.3 Cell clustering and annotation
We utilized Seurat R package (v4.4.0) and followed its pipeline for downstream analyses. The filtered scRNA-seq data were normalized and scaled using a linear regression model with the “Log-normalization” method. The top 2000 highly variable genes were identified through the “FindVariableFeatures” function. We then applied the “Harmony” package to correct for batch effects between samples and reduced dimensionality using Principal Component Analysis (PCA). Clustering of cells was performed with the “FindClusters” function, setting the resolution at 0.6. The dimensionality of each dataset was reduced using UMAP. Cell type identification was performed using the Signature-based Cell Type Annotation (SCTA) method, following the Hierarchical Cell Annotation pipeline developed by Yang et al. [4]. This novel approach integrates cells’ characteristic expression patterns with established canonical marker genes to achieve accurate and hierarchical classification of major and minor cell populations.
2.4 Consensus non-negative matrix factorization (cNMF)
GeneNMF R package (v0.9.1, https://github.com/carmonalab/GeneNMF) was used to infer gene expression programs from scRNA-seq data. The K-value was set as 6 to 10. The represented genes of distinct programs were subjected to an enrichment analysis using the “fgsea” R package (v1.30.0). Based on the enrichment results, six metaprograms (MPs) were identified and the remaining one was defined as a mixed program [27,28,29,30].
2.5 Metabolic activity analysis
Comparative analyses of metabolic activity across different groups were performed using the MetabolismExplorer R package (v0.1.0) for bulk RNA-seq datasets and the scMetabolism R package (v0.2.1) for single-cell RNA-seq datasets, respectively [31]. We followed the detailed instruction of the aforementioned R packages on github (scMetabolism: https://github.com/wu-yc/scMetabolism, MetabolismExplorer: https://github.com/StevenJiajunli/MetabolismExplorer, Supplementary Table S2).
2.6 Calculation of gene signature scores
For bulk RNA-seq data, the activity of each metabolic pathway was evaluated via GSVA R package (v1.46.0) using the Z-score transformation, defined as the mean expression of all genes in a given pathway normalized by their standard deviation across samples [32]. In terms of scRNA-seq data, individual cells were scored using the AddModuleScore function, which calculated the average expression levels of selected genes at the single-cell level and subtracted by the aggregated expression of control feature sets [33].
2.7 Empirical cumulative distribution analysis
Empirical cumulative distribution curves (eCDFs) were generated using the “stat_ecdf” function from the ggplot2 package in R. Distributions were plotted by group, with stepwise curves representing the cumulative fraction of values.
2.8 Differential analysis
For bulk RNA-seq data, differential expression analysis was performed using the limma R package (v3.54.2). Genes with a FDR < 0.05 and an absolute log2fold change > 1 were considered significantly differentially expressed. While for scRNA-seq data, differential expression analysis was conducted using the Seurat R package (v4.4.0). Cells were grouped by defined biological conditions, and differentially expressed genes (DEGs) between groups were identified using the likelihood-ratio test implemented in the “FindMarkers” function. The analysis was parallelized using the future R package (v1.31.0) to improve computational efficiency. Genes expressed in at least 25% of cells within either group, exhibiting a FDR < 0.05 and absolute log2fold change > 0.5 were considered significant.
2.9 High dimensional WGCNA (hdWGCNA)
We used hdWGCNA R package (v0.2.14) to identify HCC specific cholesterol metabolism related signatures. We followed the official pipeline released by Morabito et al., the hdWGCNA package designers, which is available on github repository (https://smorabit.github.io/hdWGCNA/) [18].
2.10 Screening of core genes involved in cholesterol metabolic reprogramming of HCC
A three-step pipeline was built for screening optimal genes involved in cholesterol metabolic reprogramming of HCC. Details of the three steps are as follows:
2.10.1 Step1: Collecting genes that are associated with cholesterol metabolic reprogramming
We first stratified malignant cells into three subgroups based on their cholesterol scores: LCholesterol (Score < − 0.09676, bottom 25%), DTCholesterol (− 0.09676 ≤ Score ≤ 0.14087, 25% to 75%), and HCholesterol (Score > 0.14087, top 25%). Then, we used single cell differential analysis (HCholesterol versus LCholesterol) and hdWGCNA to identify genes that was up-regulated and associated with HCholesterol subgroup, respectively. Subsequently, overlapping genes identified in the two analyses were collected and subjected to further screening.
2.10.2 Step2: Screening of core genes
With an intention to screen core genes involved, an integrative machine learning approach composed of five individual algorithms including LASSO, Boruta, GBM, RandomForest and XGBoost was utilized [34]. The malignant cells from Zhang et al.’s single cell atlas were used to screen the optimal genes involved in cholesterol metabolic reprogramming.
The glmnet R package (v4.1-8) was used to perform LASSO regression, which introduces an L1 regularization term to penalize model complexity and shrink less informative feature coefficients toward zero, thereby selecting the most predictive genes. A binomial model was applied with the regularization parameter α set to 1, corresponding to LASSO penalization. Model tuning was conducted using 10-fold cross-validation to determine the optimal regularization parameter (λ). The value of λ that minimized the cross-validation error (λ_min) was selected, and the final model was refitted using this optimal λ. Genes with non-zero regression coefficients in the final LASSO model were considered as selected features for subsequent analyses.
A random forest model was constructed using the randomForest R package (v4.7-1.2) with 1,000 trees to ensure stable estimation. Model reproducibility was ensured by setting a fixed random seed. Variable importance was evaluated using the mean decrease in Gini index, and genes were ranked according to their importance scores. The top 20 genes were considered as key contributors to cholesterol metabolism-associated phenotypes and were used for downstream analyses.
The xgboost R package (v1.7.8.1) was used to implement the XGBoost algorithm, a gradient boosting framework capable of iteratively optimizing residual errors from previous trees while incorporating regularization to prevent overfitting. Key hyperparameters included a maximum tree depth of 3, a learning rate (eta) of 0.1, and 50 boosting rounds.
The gbm R package (v2.2.2) was applied to train GBM models, which build additive learners in a forward stage-wise manner to sequentially correct residuals and refine feature importance rankings. Model parameters included 500 trees, an interaction depth of 3, and a learning rate (shrinkage) of 0.01. Five-fold cross-validation was applied to determine the optimal number of boosting iterations. Variable importance was assessed based on the relative influence of each gene, and genes were ranked accordingly. The top 20 genes identified by the GBM model were considered as important features and retained for subsequent analyses.
Finally, the Boruta R package (v8.0.0) was applied to identify all relevant features by iteratively comparing the importance of actual genes with their randomized “shadow” counterparts, thereby eliminating non-informative variables. The algorithm was run with 50 iterations, and genes confirmed or tentative by Boruta were retained as important features. These selected genes were subsequently used for downstream integrative analyses.
2.10.3 Step3: External validation
We examined whether the expression levels of selected genes varied according to the cholesterol metabolic state of the malignant cells through eCDF analysis using an external single cell cohort (GSE149614, HCholesterol versus LCholesterol malignant cells).
2.11 Survival analysis
Survival analysis was performed to evaluate the prognostic significance of candidate genes. Patients were stratified into high- and low-expression groups according to the median value of the corresponding gene or signature score. Kaplan Meier (KM) survival curves were generated using the survival (v3.7-0) and survminer (v0.4.9) R packages, and the hazard ratio (HR) and 95% confidence interval (CI) were calculated using univariate Cox proportional hazards regression.
2.12 Cell culture and establishment of stable knockdown cells
Murine hepatocellular carcinoma cell lines Hepa1-6 and Hep53.4 cells were descried in previous publication [4]. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin at 37 °C in a humidified incubator with 5% CO₂. For genetic silencing, lentiviral vectors carrying FDPS-targeting short hairpin RNA (shFDPS) or a non-targeting control (shCtrl) were obtained from GeneChem (Shanghai, China). FDPS knockdown was achieved using lentiviral particles encoding shRNA sequences targeting mouse or human FDPS mRNA. After 48 h, successfully transduced cells were selected with 2 µg/mL puromycin for 5–7 days. Knockdown efficiency was verified by quantitative RT–PCR and immunoblotting before downstream assays.
2.13 Western blot analysis
Hepatocellular carcinoma cells were transduced with lentiviral vectors expressing shCtrl or three independent shRNAs targeting FDPS (shFDPS #1, shFDPS #2, and shFDPS #3). After selection with puromycin, cells were harvested for protein extraction.
Cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail on ice for 30 min, followed by centrifugation at 12,000 × g for 10 min at 4 °C. The supernatants were collected, and protein concentrations were determined using a BCA assay.
Equal amounts of protein were resolved by SDS–PAGE and transferred onto PVDF membranes. Membranes were blocked with 5% non-fat milk in TBST for 1 h at room temperature and incubated overnight at 4 °C with a primary antibody against FDPS (abcam, ab153805) and GAPDH (proteintech, 60004-1-Ig). After washing, membranes were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using enhanced chemiluminescence (ECL), and images were captured using a digital imaging system.
2.14 RNA extraction and quantitative RT–PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized with the PrimeScript RT reagent kit (Takara), and quantitative PCR was performed using SYBR Green Master Mix (Bio-Rad) on a QuantStudio 5 Real-Time PCR System (Applied Biosystems). The relative expression of target genes was normalized to β-actin using the 2⁻ΔΔCt method. Primers for FDPS, SREBF2, HMGCR, MVK, IDI1, LDLR, INSIG1, ABCG1, CCND1, MYC, CDKN1A (p21), BCL2, and BAX were designed using Primer-BLAST and validated for specificity.
2.15 Cell proliferation assay
Proliferation dynamics were measured using the xCELLigence E-Plate 16 system. Cells (5 × 10³ per well) were seeded and monitored for up to 72 h under standard culture conditions. The impedance-based cell index was normalized at the time of seeding. Growth curves were generated automatically, and proliferation rates were calculated from the slope of the exponential phase. Each experiment was performed in triplicate and repeated independently at least three times.
2.16 Cell migration assay
Real-time migration assays were performed using the xCELLigence RTCA DP system (Agilent Technologies). Briefly, 3 × 10⁴ cells were seeded in serum-free medium in the upper chamber of CIM-Plate 16 wells, while the lower chamber contained medium with 10% FBS as chemoattractant. Cell index values were automatically recorded every 15 min for up to 30 h, and migration kinetics were analyzed using RTCA software.
2.17 Animal model construction and treatment
All animal experiments were performed in accordance with and approved Institutional Animal Careand Use Committee (lACUC). Six- to eight-week-old male C57BL/6 mice (purchased from Vital River Laboratory Animal Technology Co., Ltd.) were housed under specific pathogen–free conditions with a 12-hour light/dark cycle and ad libitum access to food and water. Hepa1-6 murine hepatocellular carcinoma cells (2 × 10⁶ cells in 200 µL PBS) were injected orthotopically into liver. When tumors reached 80–100 mm³, mice were randomized into four groups (n = 6–8 per group): (1) Saline control (vehicle, intraperitoneal injection), (2) αPD-1 (10 mg/kg, i.p., every three days), (3) Alendronic acid (1 mg/kg, i.p., every three days), (4) Combination therapy (αPD-1 plus alendronic acid, same dosing schedule). Tumor-bearing mice were anesthetized with 1.5% isoflurane and tumor volumes were measured every 4 days through high-resolution ultrasound imaging, which were calculated as (length × width²)/2. And the body weight of mice was recorded concurrently.
2.18 Flow cytometry
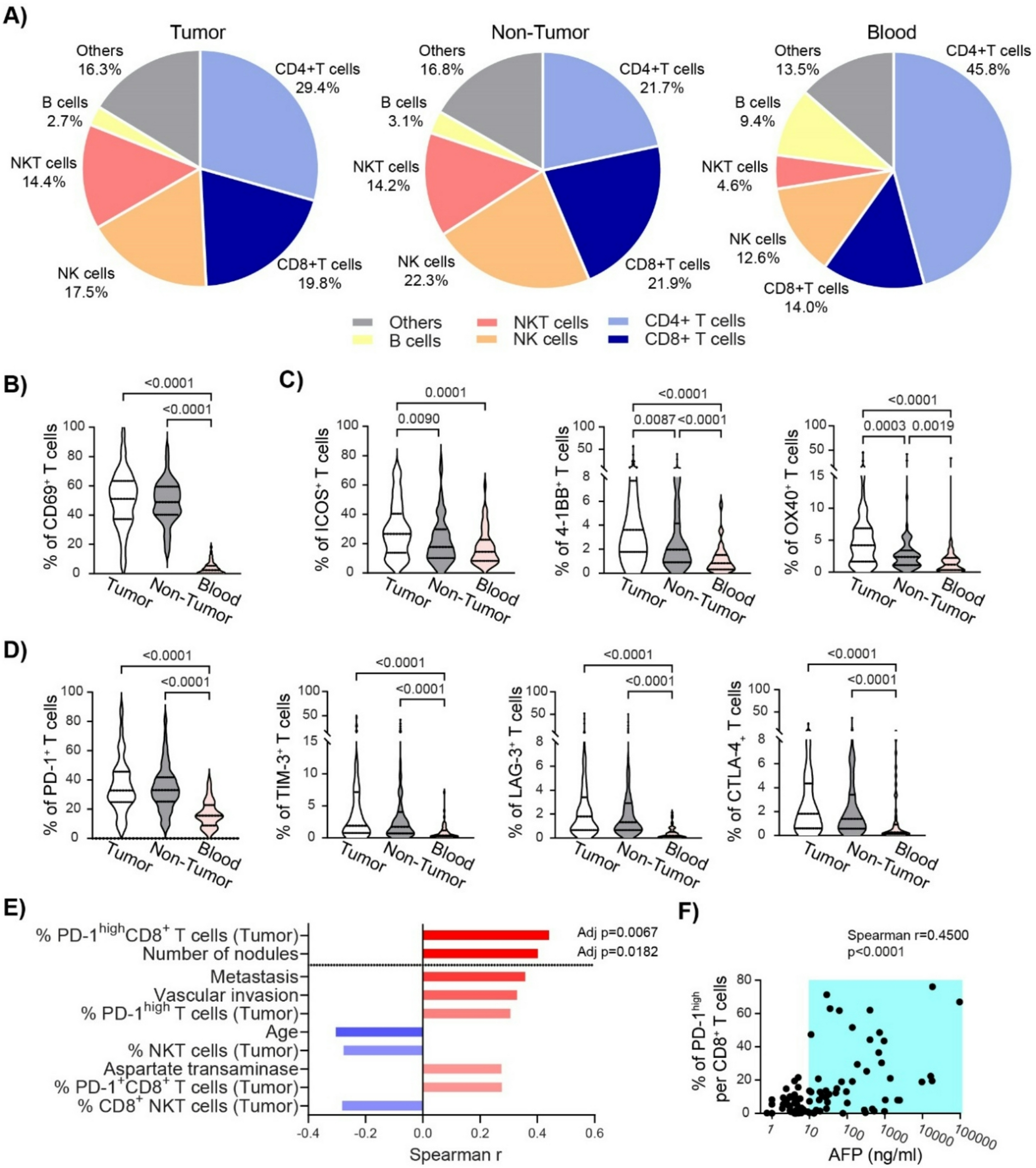
Tumors were excised from mice, minced into small fragments, and enzymatically digested in RPMI-1640 containing collagenase and DNase I at 37 °C with gentle agitation. The resulting cell suspension was filtered through a 70 μm cell strainer to obtain single-cell suspensions. Red blood cells were lysed using ACK lysis buffer when necessary. Cells were washed with DPBS and resuspended in staining buffer (DPBS supplemented with 1% FBS). For surface staining, cells were incubated with the appropriate antibodies at room temperature for 30 min, followed by a wash with DPBS. A fixable viability dye (FVD780-eFluor™ 780, eBioscience, 65-0865-14) was used to exclude dead cells. For intracellular staining, cells were fixed and permeabilized using the Fixation/Permeabilization Kit (BD Biosciences, 554714) according to the manufacturer’s instructions, followed by antibody staining in permeabilization buffer. The following anti-mouse antibodies were used: CD45-BUV395 (BD Biosciences, 564279), CD3-AF488 (BioLegend, 152322), CD4-PE/Cyanine5 (BioLegend, 100410), CD8a-BV650 (BioLegend, 100742), CD8a-BV785 (BioLegend, 100750), CD11b-BV510 (BioLegend, 101263), Ly6C-BV605 (BioLegend, 128035), F4/80-BV605 (BioLegend, 743281), Ly6G-AF700 (BD Biosciences, 561236), CD19-APC (BioLegend, 152409), NK1.1-PE (BioLegend, 108707), and CD11c-PE/Cyanine7 (BioLegend, 117317). Flow cytometry data were acquired on a BD LSRFortessa cytometer and analyzed using FlowJo software (BD). Immune cell populations were gated on live singlets and CD45⁺ leukocytes prior to lineage-specific marker analysis.
2.19 Statistical analysis
Statistical analyses were performed using GraphPad Prism (v12.0) for experimental data and R (v4.2.1) for sequencing data and associated clinical variables. Comparisons between categorical variables were conducted using the χ² test or Fisher’s exact test, while comparisons of continuous variables were performed using Student’s t-test, Wilcoxon rank-sum test, or one-way ANOVA, as appropriate. Paired t-tests were used for paired comparisons. Survival analyses were performed using the log-rank test. A P value < 0.05 was considered statistically significant. Unless otherwise specified, all experiments were independently repeated at least three times.




Comments (0)