
Remember me
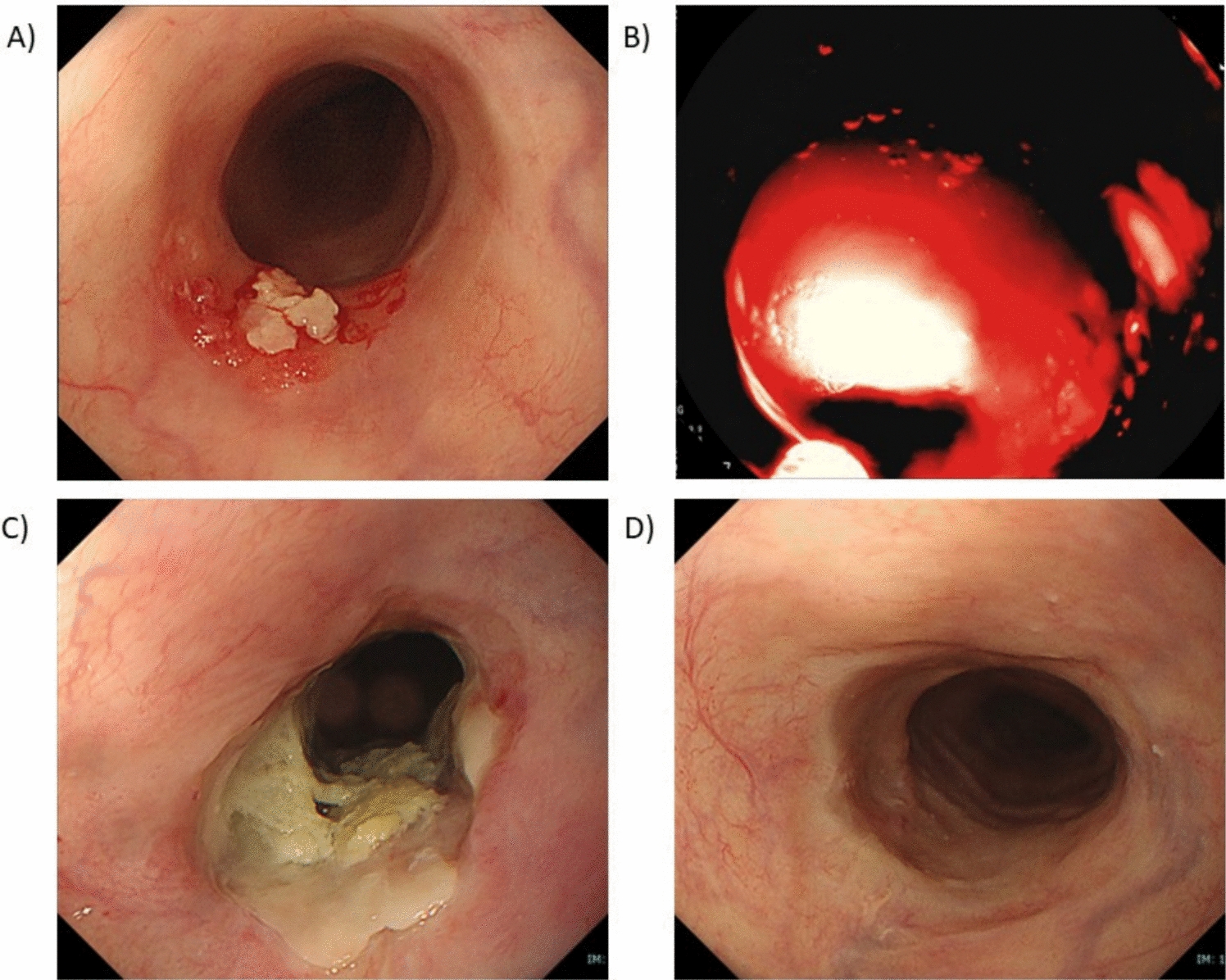
In addition to the 41 TRPV6 variants previously reported to be functionally impaired based on Ca2+ imaging assays [11,12,13,14,15], we identified four nonsynonymous TRPV6 variants of unknown functional consequence (Table 1). The c.715_724del [p.Val239SerfsTer53] variant was detected in two German, one French, and one Polish patients with pancreatitis; the c.1137C>A [p.Tyr379Ter] variant was identified in a Chinese patient; the c.1759_1761del [p.Tyr587del] variant in a Japanese patient; and the c.1870C>T [p.Arg624Ter] variant in two Austrian, one French, and one Japanese patient. In addition, six splice-site variants were identified: c.347-2A>G, c.469+1G>C, c.1029+1G>A, and c.1407-2A>G variants in French patients (one each), c.607+5G>C in a Japanese patient, and c.2015+2T>C in a Chinese patient.
Table 1 Non-synonymous TRPV6 variants detected in patients in this studyTRPV6 activity was impaired in the presence of the novel nonsynonymous TRPV6 variantsTo assess the functional impact of the nonsynonymous TRPV6 variants described above, we performed Ca2+ imaging assays. The nonsense variant c.1137C>A [p.Tyr379Ter] was excluded, because it was predicted to abolish the Ca2+ pore domain essential for TRPV6 function [16]. Compared with cells expressing wild-type TRPV6, HEK293 cells expressing TRPV6 variants showed a significantly reduced increase in intracellular Ca2+ concentration ([Ca2+]i) (Supplementary Fig. 1).
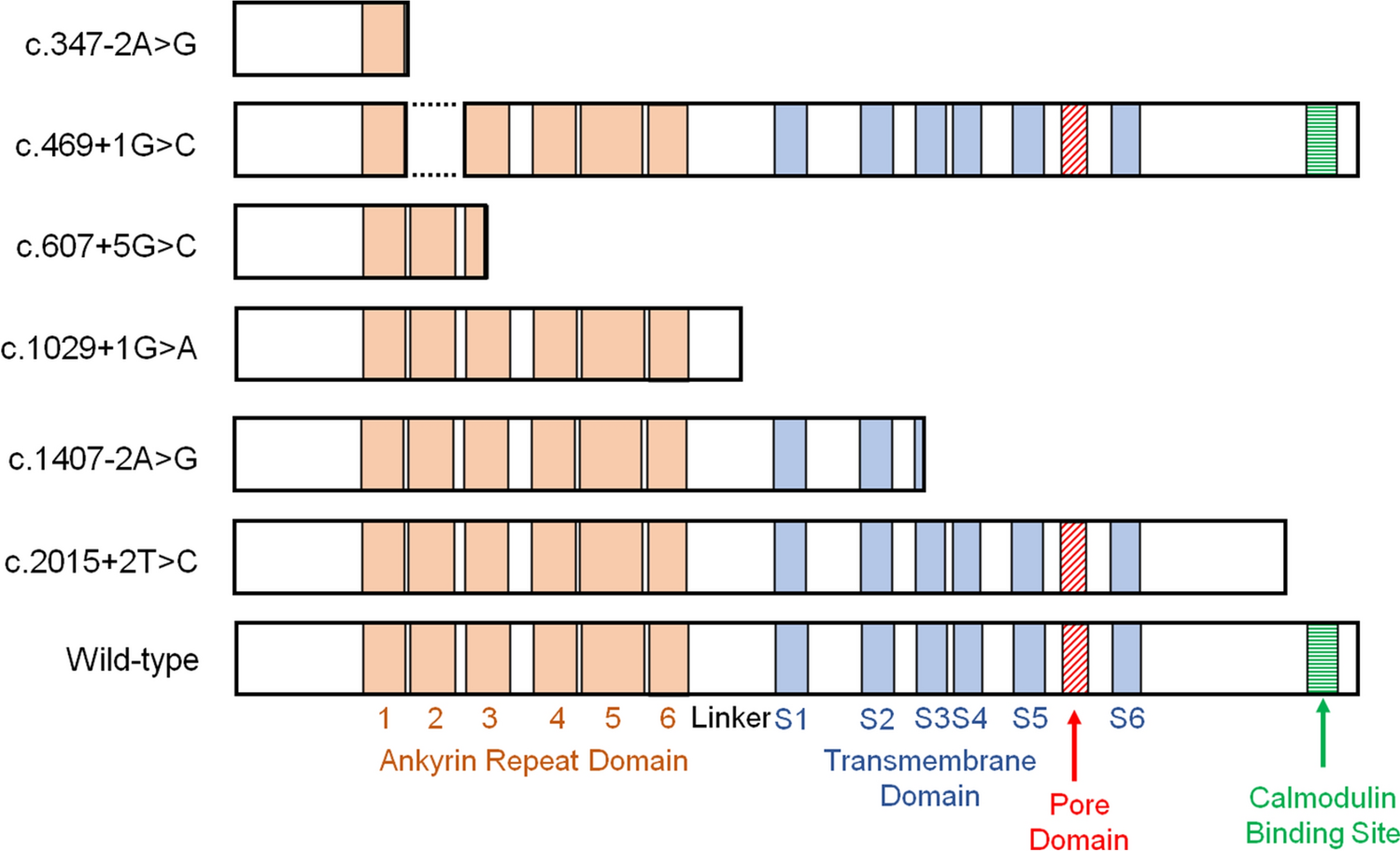
Minigene assayWe performed minigene assays to evaluate the splicing consequences of the splice-site variants (Table 2, Supplementary Figs. 2–7). Reverse transcription PCR of the wild-type construct generated a 457 bp fragment, whereas amplification of the c.347-2A>G construct yielded a larger 490 bp fragment due to retention of the first 33 bp of intron 2 before the exon 3 splice donor. This was predicted to result in the insertion of three new amino acids followed by a premature stop codon. The c.469+1G>C variant was predicted to cause skipping of the entire exon 3, resulting in the deletion of 41 amino acids and loss of the ANK2 domain. The c.607+5G>C variant was predicted to cause the skipping of the last 91 bp of exon 4, leading to a frameshift and early termination within the ANK3 domain. The c.1029+1G>A variant was predicted to include the entire intron 7, causing a frameshift and termination in the linker region. The c.1407-2A>G variant was predicted to cause complete exon 11 skipping, resulting in termination at amino acid 470. These five variants caused the loss of the Ca2+ pore domain. The c.2015 + 2T>C variant was predicted to include the first 17 bp of intron 14, generating 40 novel amino acids and the loss of the calmodulin binding site. Because loss of this site diminishes TRPV6 activity [11], the c.2015 + 2T>C variant was considered functionally impaired. Collectively, all six splice-site variants were predicted to impair TRPV6 function and to be pathogenic [35]. The predicted structural consequences based on the minigene assays are summarized in Fig. 1.
Table 2 Predicted splicing outcome of splice-site variants based on minigene assaysFig. 1
Predicted structures of the mutated TRPV6 protein based on the minigene assays. Normal splicing of the wild-type sequence and aberrant splicing of the mutant sequence are illustrated for each variant
Overview of the patients carrying functionally impaired TRPV6 variantsWe aimed to clarify the clinical characteristics of patients carrying functionally impaired TRPV6 variants. A total of 45 nonsynonymous variants (41 previously reported and four identified in this study) and six splice-site variants were included. In total, 94 patients with alcohol-unrelated pancreatitis (70 with CP and 24 with RAP) were retrospectively analyzed (Table 3), comprising 33 French, 20 Japanese, 18 Chinese, 11 German, 7 Polish, 3 Indian, and 2 Austrian patients. These patients are collectively referred to as having TRPV6-related pancreatitis. Of the 94 patients, 54 (57.4%) were male. Thirty-two (34.0%) had a family history of pancreatitis and were classified as hereditary/familial, while 62 (66.0%) were classified as idiopathic. The median age at the final follow-up was 26 years. Two patients carried homozygous TRPV6 variants (p.Arg174Ter and p.Arg646Trp, one each). Although no patients were compound heterozygous for functionally impaired TRPV6 variants as defined herein, eight patients had other nonsynonymous TRPV6 variants (p.Ala210Val/p.Asp324Asn, p.Ile223Thr/p.Leu392Phe, p.Leu392Phe/p.Gly451Glu, p.Ile223Thr/p.Arg425Gln, p.Ile223Thr/p.Gly428Arg, p.Ile223Thr/p.Ile580Phe, p.Val492Leu/p.Ala626Pro, and c.607+5G>C/p.Ile223Thr, all one each). Twenty-one (22.3%) patients were double heterozygous for pancreatitis risk variants in other susceptibility genes, including 14 with SPINK1 (8 with p.Asn34Ser and 6 with c.194+2T>C), 2 with CTRC (both p.Arg254Trp), and 5 with CFTR (3 with p.Phe508del and 2 with p.Arg117His) [36]. No patients carried pathogenic PRSS1 variants (p.Arg122His or p.Asn29Ile).
Table 3 Clinical characteristics of enrolled patientsOnset of symptomsMost patients with TRPV6-related pancreatitis developed symptoms by the age of 30 (Fig. 2A, Supplementary Fig. 8A). The median age at symptom onset was 16 years. Twelve patients (12.8%), 21 (22.3%), 64 (68.1%), and 76 (80.9%) developed symptoms by the ages of 5, 10, 20, and 30, respectively. Among the 89 symptomatic patients, the initial presentation was acute pancreatitis in 67 patients, abdominal pain in 14, abdominal and back pain in 6, and vomiting in 2 patients. We compared the age at symptom onset in patients with TRPV6-related pancreatitis to those with PRSS1-related pancreatitis (p.Arg122His and p.Asn29Ile; n = 68), SPINK1-related pancreatitis (p.Asn34Ser, p.P45Ser, and c.194+2T>C; n = 90) [37], and PV-negative pancreatitis (n = 314) (Table 3, Supplementary Table 8). The median age at symptom onset (95% CI) was 8 years (4.0–12.0) in PRSS1-related pancreatitis, 14 years (11.5–16.5) in SPINK1-related pancreatitis, and 32 years (27.1–36.9) in PV-negative pancreatitis. The age at symptom onset in TRPV6-related pancreatitis was significantly younger than that in PV-negative pancreatitis (P < 0.001) but older than that in PRSS1-related pancreatitis (P = 0.001). No significant difference was observed between TRPV6- and SPINK1-related pancreatitis.
Fig. 2
Comparison of different pathogenic genotypes on clinical outcomes. Kaplan–Meier curves showing the cumulative rates of A symptom onset, B pancreatic calcification, C pancreatic exocrine insufficiency, and D diabetes mellitus, according to genotype (TRPV6-related, PRSS1-related, SPINK1-related, or PV-negative pancreatitis). Censored subjects are indicated on the Kaplan–Meier curves by tick marks
Pancreatic calcificationForty-nine patients (52.1%) with TRPV6-related pancreatitis were diagnosed with pancreatic calcification, with a median age of 29 years (Fig. 2B, Supplementary Fig. 8B). The cumulative incidence rates of pancreatic calcification were 19.4%, 55.5%, 74.6%, and 81.5% at the ages of 20, 30, 40, and 50 years, respectively. The age at diagnosis was significantly younger in patients with TRPV6-related pancreatitis than in those with PV-negative pancreatitis (P < 0.001). No significant differences were observed when compared with patients with PRSS1-related or SPINK1-related pancreatitis.
PEI and DMTwenty-one patients (22.3%) and 15 patients (16.0%) with TRPV6-related pancreatitis were diagnosed with PEI and DM, respectively (Fig. 2C, D, Supplementary Fig. 8C, D). The cumulative rates of PEI were 4.9%, 20.1%, 40.8%, and 49.6%, and those of DM were 2.9%, 10.8%, 39.3%, and 45.4% at the ages of 20, 30, 40, and 50, respectively. The age at diagnosis of PEI and DM was significantly younger in patients with TRPV6-related pancreatitis compared to those with PV-negative pancreatitis (P < 0.001 for PEI and P = 0.006 for DM). In contrast, the age at diagnosis of PEI was older in TRPV6-related pancreatitis patients than in PRSS1-related pancreatitis (P = 0.002), while no significant difference was observed for DM. There was no significant difference in the age at diagnosis of either PEI or DM between patients with TRPV6-related and SPINK1-related pancreatitis.
InterventionsAmong the 94 patients with TRPV6-related pancreatitis, 37 (39.4%) underwent intervention: endoscopic treatment alone in 30 (81.1%), surgery alone in 4 (4.3%), a step-up approach in 2 (2.1%), and a top-down approach in 1 (1.1%). The median ages at first endoscopic treatment and at first intervention (either endoscopic or surgical) were 37 and 35 years, respectively (Fig. 3A–C, Supplementary Fig. 9A–C). The cumulative rates of all interventions were 16.7%, 41.6%, 58.0%, and 69.9% at ages 20, 30, 40, and 50 years, respectively. For endoscopic treatment, the cumulative rates were 15.4%, 35.4%, 51.2%, and 62.6% at ages 20, 30, 40, and 50 years, respectively. For surgery, the cumulative rates were 2.5% and 12.0% at ages 20 and 30 years, respectively. The age at first intervention was significantly younger in patients with TRPV6-related pancreatitis than in those with PV-negative pancreatitis (P < 0.001), whereas no significant differences were observed compared with patients with PRSS1- or SPINK1-related pancreatitis. Similar results were observed for endoscopic treatment (P < 0.001 vs. PV-negative, P = 0.21 vs. PRSS1, P = 0.12 vs. SPINK1). In contrast, patients with TRPV6-related pancreatitis underwent their first procedure at an older age than those with PRSS1-related pancreatitis (P = 0.002), whereas no significant differences were observed compared with patients with SPINK1-related or PV-negative pancreatitis.
Fig. 3
Timing of the first interventions for pancreatitis and the diagnosis of pancreatic cancer according to pathogenic genotypes. Kaplan–Meier curves showing the cumulative rates of A endoscopic treatment, B surgery, C all interventions, and D pancreatic cancer diagnosis according to genotype (TRPV6-related, PRSS1-related, SPINK1-related, or PV-negative pancreatitis). Censored subjects are indicated on the Kaplan–Meier curves by tick marks
Diagnosis of pancreatic cancerPancreatic cancer was not observed in patients with TRPV6-related pancreatitis. In contrast, it was diagnosed in one patient with the PRSS1 p.Arg122His variant, one patient with the SPINK1 p.Asn34Ser variant, and eight patients with PV-negative pancreatitis (Fig. 3D, Supplementary Fig. 9D).
Subgroup analysis excluding double-heterozygous patientsAmong the 94 patients with TRPV6-related pancreatitis, 14 were double heterozygous for TRPV6 and SPINK1 variants. Therefore, a subgroup analysis was conducted on the remaining 80 patients (Supplementary Tables 9 and 10; Supplementary Figs. 10 and 11). All significant differences observed in the overall cohort (n = 94) remained statistically significant in this subgroup. In this subgroup, the ages at diagnosis of pancreatic calcification (P = 0.03) and at first endoscopic treatment (P = 0.041) were significantly higher than those in patients with SPINK1-related pancreatitis.
Caerulein-induced pancreatitis was exacerbated in pancreas-specific Trpv6 knockout miceFinally, we investigated whether deletion of Trpv6 in the pancreas affects the progression of pancreatitis in mice. To this end, we established pancreas-specific Trpv6 CKO mice (Supplementary Fig. 12). A shorter PCR band was detected in pancreatic genomic DNA of Trpv6 CKO mice, indicating Cre-mediated recombination, whereas other organs (lung, heart, liver) showed no evidence of recombination. Neither the floxed nor CKO mice exhibited significant histological abnormalities in the pancreas up to 90 days of age, similar to the Trpv6mut/mut mice [11]. No PanIN formation was detected in Trpv6 CKO mice, even at 180 days of age (data not shown).
We induced pancreatitis by eight repetitive injections of caerulein over two consecutive days (Fig. 4A–D). Serum amylase levels increased 8 h after the first caerulein injection, decreased at 24 h, and elevated again at 32 h. Serum amylase levels were significantly higher in Trpv6 CKO mice than in control floxed mice at 8 h. Histologically, Trpv6 CKO mice developed more severe pancreatitis, as shown by more severe pancreatic edema, inflammatory cell infiltration, and acinar necrosis, compared to Trpv6 floxed mice (Supplementary Fig. 13). On day 5, pancreatic fibrosis was more evident in Trpv6 CKO mice, as assessed by Sirius Red staining.
Fig. 4
Caerulein-induced pancreatitis was exacerbated in pancreas-specific Trpv6 conditional knockout mice. A–D Trpv6 floxed mice and pancreas-specific Trpv6 CKO mice received eight hourly intraperitoneal injections of caerulein (100 μg/kg body weight) or saline for two consecutive days. Blood samples were collected at 0 h (just before the first caerulein injection), 8 h, 24 h, and 32 h, and mice were euthanized at either 32 h or 96 h. A Scheme of the experiments. B Blood samples were obtained at the indicated time points, and serum amylase levels were measured. C Representative H&E staining of the pancreas at 32 h. Scale bar = 100 μm. D Representative H&E staining and Sirius Red staining of the pancreas at 96 h after the first caerulein injection. E, F CP was induced by 6-hourly intraperitoneal injections of caerulein (100 μg/kg body weight), 3 days/week for four consecutive weeks. E Scheme of the experiments. F Representative H&E staining and Sirius Red staining of the pancreas after 4 weeks
CP was induced by six intraperitoneal injections of caerulein, administered 3 days/week for four consecutive weeks (Fig. 4E). Histological analysis revealed that Trpv6 CKO mice exhibited more severe pancreatitis than floxed controls, characterized by increased inflammatory cell infiltration, acinar cell loss, and fibrosis (Fig. 4F). These findings indicate that pancreatic Trpv6 plays a protective role in both acute and chronic pancreatitis in mice.
Effects of Trpv6 deletion on pancreatic organoidsTRPV6-related pancreatitis has been proposed as a “channelopathy” that impairs ductal secretion [11, 38], based on its predominant expression in human pancreatic duct cells identified by single-cell transcriptome profiling [39]. To investigate the effect of Trpv6 deletion on ductal cells, we generated pancreatic organoids from Trpv6 floxed and Trpv6 CKO mice (Supplementary Fig. 14). These organoids expressed ductal markers such as Slc9a1 and Slc4a4, whereas acinar markers such as Prss1 and Spink1 were undetectable, confirming their ductal phenotype.
Treatment with forskolin increased organoid size, indicating enhanced fluid secretion into the lumen [34]. However, the relative increase in organoid size after forskolin stimulation did not differ significantly between Trpv6 floxed and Trpv6 CKO organoids. Similarly, both groups showed comparable growth and morphological responses following Ca2+ treatment at various concentrations. These findings suggest that Trpv6 deletion does not significantly affect the function or phenotype of pancreatic ductal organoids.
TRPV6 expression is upregulated in pancreatic acinar cells in response to caerulein treatmentWe hypothesized that TRPV6 is upregulated in pancreatic acinar cells following pancreatitis stimuli and may play a protective role against pancreatitis. Immunohistochemical staining revealed increased TRPV6 expression at 32 h after caerulein treatment in Trpv6 floxed mice, whereas no such increase was observed in Trpv6 CKO mice (Supplementary Fig. 15). In addition, repeated intraperitoneal injections of caerulein elevated Trpv6 mRNA expression in the pancreas of male C57BL/6J mice. These findings indicate that TRPV6 is upregulated in pancreatic acinar cells in response to caerulein stimulation.
Comments (0)