
Remember me




As already mentioned, the compounds produced by Actinobacteria have a long history of use in the treatment of TB. From this great asset, many molecules with antimycobacterial activity were discovered. However, the activity of the vast majority of these molecules was not explored clinically, or even in animal tests. In this context, the present work provides an overview of natural substances with antimycobacterial activity derived from Actinobacteria. This review, that compiles 171 molecules with antimycobacterial activity, was performed thorough in the PubMed database using the terms: “Actinobacteria", "Actinomycete", "Streptomyces" and "Mycobacterium tuberculosis”. Three hundred eighty-four articles were retrieved, after removing duplicates and articles that not present elucidated chemical structures, 60 articles published between 1972 to 2024 were elected to deep analyses. Figure 2 depicts a bibliometric data extracted from the selected articles.
Fig. 2
Bibliometic data and Actinobacteria-producing antimycobacterial metabolites source. A Number of studies conducted on the isolation of antimycobacterial metabolites by country. B VOSviewer network visualization of the selected studies. C Distribution of actinobacteria-derived metabolites with antimycobacterial properties based on bacterial habitat (isolation source)
Of the sixty articles retrieved, the most were from Asia (68.3%), with China, Japan and Thailand having the largest contributions (29 publications, 48.3%) (Fig. 2A). Figure 2B displays the geographical distribution of the selected publications and the connections between different nations. The analysis was performed in the VOSviewer software. VOSviewer is a widely used tool for bibliometric and network analysis, enabling clear and interactive visualization of scientific data [68]. Thus, the figure helps identify the main collaborating countries and their impact on the scientific production related to the topic reviewed in this work.
The profile displayed in Fig. 2A, B demonstrates a significant concentration of scientific output in Asia, underscoring the region's leading role in research and development of novel antimicrobial agents derived from Streptomyces. These findings are consistent with those of Leite et al. [69], who identified Asia as the region with the highest number of patents related to bioactive compounds produced by Streptomyces species with antimicrobial activity. This could be related to public policies that foster technological innovation, alongside a well-established tradition in industrial microbiology and microbial bioprospecting [69].
Although microbial diversity is globally distributed, scientific advances in Actinobacteria research are largely concentrated in countries with robust infrastructure and funding. China has become a global leader in scientific output due to strategic investments in science and technology, particularly in biotechnology and public health [70]. Similarly, Japan’s prominence stems from a strong post-war tradition in antibiotic discovery and the work of key research institutions dedicated to novel antimicrobials [69].
5.2 Ecological origins of actinobacteria-derived metabolitesRegarding the sources of isolation of antimicrobial-producing actinobacteria, the presence of these microorganisms was observed in diverse environments, with emphasis on soil (40%), marine sediments (23%), plants (10%) and, animals (8%) (Fig. 2B). This can be attributed to the high complexity of terrestrial environments [71]. However, in recent decades, a growing number of studies have shown that obtaining new species of Streptomyces from terrestrial environments has become progressively more limited, with the rediscovery/reisolation of secondary metabolites already known to be produced by these bacteria being frequent [72]. In this context, actinobacteria from marine ecosystems have stood out for their remarkable biosynthetic potential. This chemical diversity is strongly influenced by the adverse physical–chemical conditions of these habitats, including extreme variations in pressure, salinity, light, and temperature [73, 74]. Furthermore, bioactive compounds obtained from marine microorganisms have proven to be particularly promising, due to the greater probability of presenting novel chemical structures and relevant pharmacological activities [75].
Several microbial communities have been identified within the internal tissues of plants as endophytes, which perform ecologically significant functions in the plant environment, being recognized for their ability to produce compounds that promote plant growth, act as repellents against insects and phytopathogens, and contribute to the tolerance of adverse abiotic stress conditions [76, 77]. In addition, actinobacteria have increasingly been recognized for their symbiotic associations with eukaryotic hosts, being widely distributed both on the external surfaces and in the digestive tract of various animals. Interestingly, certain insects show a functional dependence on these bacteria for nutritional supplementation, highlighting their ecological and functional importance in mutualistic relationships [6, 7]. Thus, actinobacteria continues to represent a promising yet underexplored reservoirs of bioactive compounds.
5.3 Classification and overview of antimycobacterial metabolites from actinobacteriaRegarding antimycobacterial potential of the retrieved metabolites, the compounds were classified based on the minimum inhibitory concentration (MIC) values, being categorized as follows: compounds with MIC > 10.0 μM were considered to have weak activity, those with MIC between 1.0 μM and 10.0 μM were classified as having moderate activity, while compounds with MIC < 1.0 μM were classified as potent. All the substances analyzed are listed in Tables 1, 2, 3. The compiled data presents the Actinobacteria specie, isolation source, local/country of isolation, and MIC, in μg/mL and μM. The chemical structures were drawn in the ChemDraw software 23.1.2 and are presented in Figs. 3, 4, 5, 6, 7, 10, 11. For the purposes of in-depth analysis and discussions, we selected only the compounds that demonstrated moderate and potent activity, since these may have greater pharmacological relevance in the context of the development of new antitubercular agents.
Table 1 NRP from Actinobacteria with antimycobacterial activityTable 2 Antimycobacterial Polyketides Isolated from ActinobacteriaTable 3 Antimycobacterial metabolites from Actinobacteria: Nucleotides, aminoglycosides, and miscellaneous compoundsFig. 3
Chemical structures of antimycobacterial NRP metabolites
Fig. 4
Chemical structures of antimycobacterial NRP metabolites
Fig. 5
Chemical structures of antimycobacterial NRP metabolites
Fig. 6
Chemical structures of PKS-derived antimycobacterial metabolites
Fig. 7
Chemical structures of PKS-derived antimycobacterial metabolites
5.3.1 Peptide antibioticsThe non-ribosomal peptide synthesis (NRPS) pathway is mediated by large multifunctional enzymes and is characterized by the production of non-ribosomal peptides (NRP), which are not encoded by genes and are not restricted to the 20 conventional amino acids. Non-ribosomal peptide synthetases (NRPS) use proteinogenic and non-proteinogenic amino acids as building blocks for the peptide chain. As a result, the products generated by this pathway present a great structural diversity and a broad spectrum of biological activities, making them valuable for application in agriculture and medicine [78,79,80]. Some common characteristics among NRP include their highly specific structure, which ensures bioactivity through a precise orientation, essential to interact with a molecular target; in addition, macrocyclization is an important characteristic, in which distant parts of the linear peptide precursor are covalently linked to each other [81]. Table 1 presents NRP isolated from different Actinomycetota species, along with their respective MIC values against mycobacterial strains.
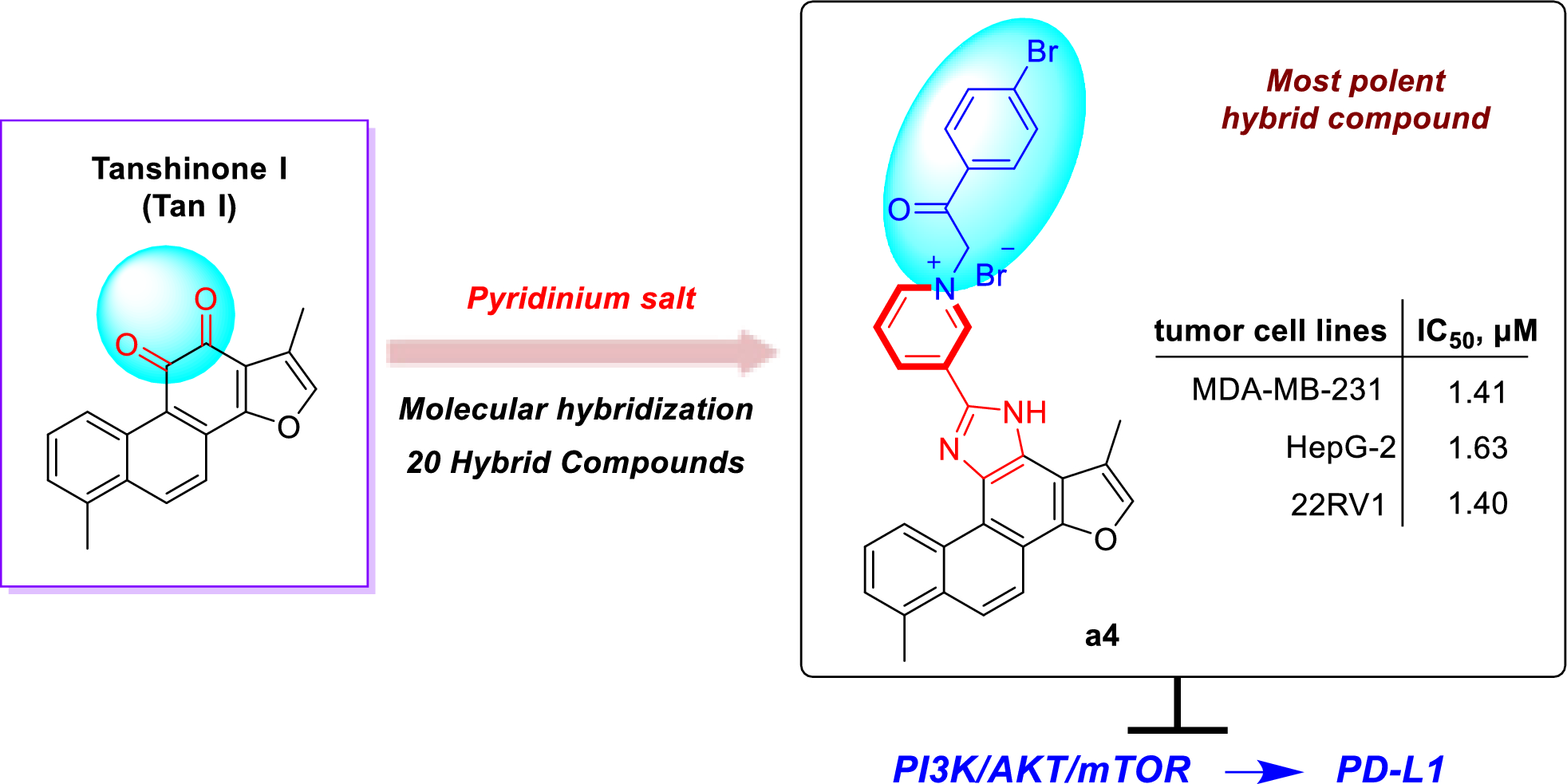
Actinomycins. Actinomycins are chromogenic cyclic peptides, first discovered in 1940 [108]. These substances stand out for being antibiotics and anticancer agents, isolated from several species of Streptomyces, of which, to date, about 30 natural and synthetic analogues have been discovered [109, 110]. These actinomycins are composed of a chromophore group and two pentapeptide chains, whose amino acid composition varies (Fig. 3).
Our analysis revealed that actinomycins exhibiting antitubercular activity were isolated from five distinct Streptomyces species, including S. griseoruber, S. smyrnaeus, S. avermitilis, and two unclassified Streptomyces species. Praveen and Tripathi [82] evaluated the actinomycin D activity against Mtb H37Rv, reporting a MIC value of 0.62 µM. Qureshi et al. [83] isolated both actinomycin D and actinomycin X2, which exhibited MIC values of 2.08 µM and 1.43 µM, respectively, against Mtb H37Rv. Similarly, Chen et al. [84] reported the isolation of actinomycin D, actinomycin X2, and actinomycin X0β. These compounds displayed MIC of 6.37 µM, 0.79 µM, and 6.29 µM, respectively, against Mtb H37Rv. In another study, Shah et al. [85] isolated actinomycin D, actinomycin X2, and actinomycin C3. These compounds demonstrated particularly potent activity against Mtb H37Rv, with MIC of 0.05 µM, 0.03 µM, and 0.03 µM, respectively. Rakhmawatie et al. [86] also reported the isolation of actinomycin D, which exhibited an MIC of 0.62 µM.
Structurally, actinomycins X2 and X0β are closely related to actinomycin D, with variation arising from the substitution of the proline residue in the polypeptide ring. Replacement by 4-trans-hydroxyproline or 4-oxo-proline yields actinomycin X0β and X2, respectively [111] (Fig. 3). In contrast, actinomycin C3 contains modifications in the D-valine residues of the polypeptide ring, which are replaced by D-allo-isoleucine [111] (Fig. 3). Among these compounds, actinomycin D is the most studied analog and continues to be widely used in the treatment of various types of cancer, especially in pediatric oncology and has been gaining prominence for its antimicrobial potential [112].
From a biochemical point of view, actinomycin D's mechanism of action is based on its ability to intercalate between GC base pairs, preventing the progression of RNA polymerase and, blocking gene transcription [113, 114]. Despite their high bioactive potential, the toxicity associated with actinomycins still represents a major obstacle to their wider clinical use [115]. However, given the global crisis of antimicrobial resistance and the urgent need for new anti-TB therapies, the re-evaluation of classic molecules, such as actinomycins, has emerged as a viable strategy. The rational use of these molecules, combined with structural modification approaches, encapsulation or combination with other agents, can reduce their side effects and broaden their therapeutic applicability.
Echinomycin. Echinomycin is a cyclic depsipeptide antibiotic known for its extensive activities against bacteria and tumor cells (Fig. 3). This compound belongs to the quinoxaline family and originates from different species of Streptomyces, Echinomycin promotes DNA damage, cell apoptosis and inhibition of bacterial RNA synthesis [89]. This substance was the first DNA bisintercalator identified, having the ability to reversibly bind to the double helix in a sequence-independent fashion, inserting one or more aromatic ring groups between adjacent base pairs. Thus, echinomycin bound to DNA inhibits transcription in bacteria, chromatin condensation and DNA replication in eukaryotic organisms, leading to cell cycle arrest [116].
The study conducted by Chen et al. [89] observed that echinomycin obtained exhibited a MIC of 0.45 μM against Mtb H37Rv. However, according to Gade et al. [117], despite its potent antimicrobial activity, echinomycin is not used clinically due to solubility and toxicity concerns. The study of Foster et al. [118] investigated the toxicity of echinomycin in mice and Beagle dogs through intravenous injections administered over five consecutive days, and the primary toxic effects were observed in the gastrointestinal, hepatic, and lymphoreticular systems.
Cyclomarins. Cyclomarins A and C are heptapeptide cyclopeptides biosynthesized via the NRPS, sharing a similar central structure but presenting crucial differences in their side chains (Fig. 3). Cyclomarin A demonstrated high activity against the virulent strain of Mtb, with a MIC of 0.48 µM [87] while cyclomarin C was capable of inhibiting Mtb H37Ra with a MIC of 0.10 µM [88].
The main distinction between these two compounds lies in the chemical modification of the hydroxylated tryptophan residue. Cyclomarin A contains an epoxide group on the side of this residue, specifically an N-(1,1-dimethyl-2,3-epoxypropyl) substitution [119, 120]. This structural difference has direct implications for the bioactivity of the compounds. Studies have shown that the presence of the epoxide group in cyclomarin A is essential for its potent activity against Mtb, since this functional group is involved in the interaction with the target protein caseinolytic protease C1 (ClpC1). In contrast, cyclomarin C, which lacks this modification, has considerably reduced antimicrobial activity, showing that small structural changes can significantly impact the pharmacological efficacy of the molecule [119].
Currently, the information available on the toxicological profile of cyclomarins is still scarce. The presence of the epoxide group in the structure of cyclomarin A, although essential for its bioactivity, represents a highly reactive functional structure that can interact non-specifically with host macromolecules, raising concerns about toxicity [119, 121]. The possibility of side effects related to cross-inhibition of human proteases or other homologous structural chaperones represents a significant limitation for the clinical use of these compounds, if a satisfactorily high therapeutic index is lacking [114, 122,123,124].
Rufomycins and ilamycins. Rufomycins and ilamycins are cyclic heptapeptides that feature an isoprenyl group attached to the nitrogen of the tryptophan ring [125] (Fig. 3). These compounds have been increasingly recognized for their potential in the treatment of TB, particularly for their inhibitory action on ClpC1, a validated and essential target for Mtb viability [126].
In the study conducted by Zhou et al. [90] eight new rufomycins compounds were evaluated for their antimycobacterial activity against a virulent strain of Mtb H37Rv. The results revealed a broad range of potencies, with MIC values ranging from 0.030 μM to greater than 10 μM. The presence of an epoxide ring in the prenyl group of tryptophan was strongly correlated with increased antimicrobial activity, while the absence of functional groups such as N-methylleucine (N-MeLeu) and m-nitro-tyrosine (m-NO₂-Tyr) resulted in the compounds losing their efficacy.
Sun et al. [91] isolated twelve ilamycins (G to R, also referred to as rufomycins), and evaluated their activity against Mtb H37Rv, yielding MIC of 0.0096–9.6 μM (Table 1). It was observed that structural modification involving reduction and cyclization at position C-33 significantly enhances the biological activity of these compounds. In contrast, oxidation at C-15 did not substantially affect activity, while oxidation at C-32 resulted in a loss of efficacy. Conversely, in another analysis, oxidation at C-15 compromised activity, whereas oxidation at C-32 contributed positively. Furthermore, the presence of a nitro group at C-43 plays a crucial role in maintaining or enhancing the observed antitubercular activity.
Although the bioactivity data is significant, authors also reported concerns about the toxicity of these compounds. Rufomycins, in particular, showed relevant cellular toxicity in murine macrophage models, with relatively low selectivity indices for some analogues, which may limit their direct clinical application without additional structural modifications [90]. These findings indicate that, although potent, these compounds may interfere with conserved cellular pathways in host cells, which calls for further investigation into their safety. Thus, the studies involving rufomycins and ilamycins reinforce the high potential of these cyclopeptides as ClpC1 inhibitors, representing a promising new class of anti-TB agents.
Tuberactinomycins. Tuberactinomycins are a family of cyclic peptide antibiotics, including viomycin and capreomycin, that exhibit potent activity against Mtb, particularly MDR and XDR strains. Structurally, they feature a unique hexapeptide core with non-proteinogenic amino acids and a guanidine group critical for ribosomal targeting [127] (Fig. 4). Their mechanism involves binding to the 30S and 50S ribosomal subunits at the interface, specifically disrupting tRNA accommodation during translocation (A- to P-site movement) and inhibiting protein synthesis.
Tuberactinomycins have historically been employed as second-line therapeutic agents in the treatment of MDR-TB [127]. However, despite their efficacy, clinical use is limited by nephrotoxicity and ototoxicity, though they remain WHO-recommended for MDR-TB when safer options (e.g., bedaquiline) are unavailable.
Viomycin was the first member of the class to be isolated and was later identified as chemically identical to tuberactinomycin B. Capreomycin, isolated from Streptomyces capreolus, was initially described as a mixture of four distinct components (capreomycins IA, IB, IIA, and IIB), which were subsequently differentiated based on structural substituents. Despite this chemical complexity, capreomycin has been clinically used as a combined formulation of all four components.
In the study by Rokuro et al. [92] tuberactinomycins A and B was isolated from the soil actinobacteria Streptomyces griseoverticillatus and inhibited Mtb ATCC 607 with MIC of 17.81 μM and 4.67 μM, respectively. Although current clinical guidelines restrict their use due to toxicity concerns, their retained efficacy against resistant strains suggests potential for revitalization. Prior research indicates that modulating culture conditions [128] or engineering BGC could yield novel analogs with improved pharmacological profiles. Semi-synthetic modifications of these ciclopeptides may further expand the therapeutic utility. Beyond TB, repurposed tuberactinomycins could address other priority pathogens, as evidenced by viomycin’s activity against other priority pathogens, as vancomycin-resistant Enterococcus (VRE) and methicillin-resistant Staphylococcus aureus (MRSA) [129, 130].
Hytramycins. Hytramycin I and V are cyclohexapeptides, each bearing three unusual piperazic acid moieties, two adjacent and the third flanked by another amino acid [93, 131] (Fig. 4). Both compounds were tested against Mtb H37Rv and exhibited MIC of 17.84 μM and 9.27 μM,
Comments (0)