
Remember me

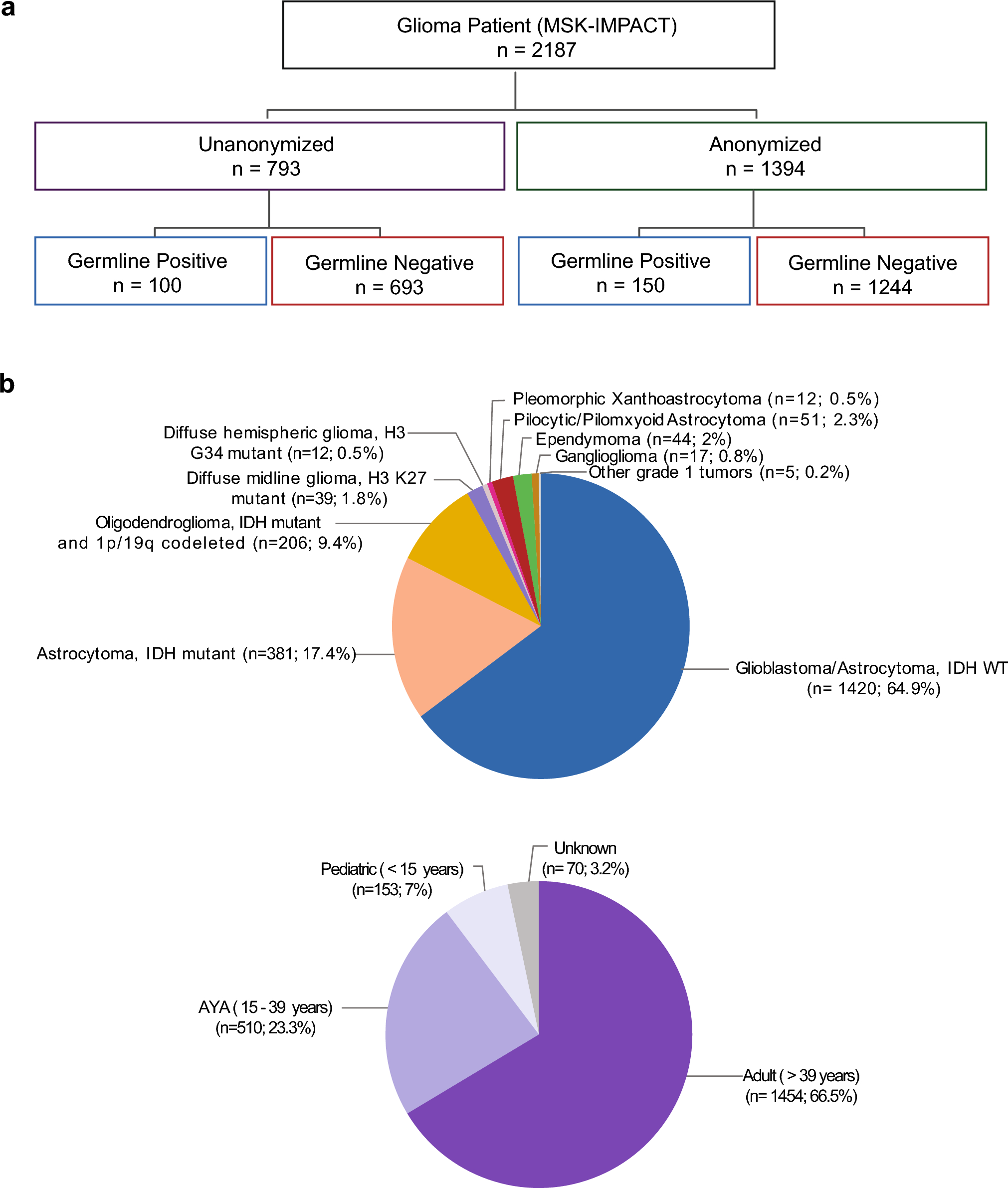





1394 patients underwent sequencing of their tumor and matched normal tissue and were anonymized prior to analysis for a P/LP germline alteration and comprise the “anonymized subset.” An additional 793 patients consented to sequencing of their tumor and clinical review of their germline sequencing data and comprise the “un-anonymized subset” (Fig. 1a).
Fig. 1
Breakdown of the study cohort. a: Analysis of germline variants was performed in an anonymized and un-anonymized subset. b: Breakdown of cohort by age and tumor type
The combined cohort comprised of 2187 patients between 0.2 and 94 years of age: 1,454 adults (> 39 years, 66.5%), 510 adolescents and young adults (age 15–39 years, 23.3%), and 153 pediatric patients (< 15 years, 7%). 64.9% had glioblastoma, IDH WT, 17.4% had an IDH mutant astrocytoma and 9.4% had an oligodendroglioma, IDH mutant and 1p/19q codeleted (Fig. 1b for lesser represented tumor types).
Frequency of germline alterationsP/LP germline alterations were identified in 11% (150/1394, 95% CI: 9.1%-12.4%) of patients in the unbiased anonymized subset and in 11% (250/2187, 95% CI: 10.1–12.8%) of the entire patient cohort (Fig. 1a, Supplementary Table 1). Of these 250 patients, 8 patients harbored a germline TP53 mutation and had Li-Fraumeni syndrome—a prevalence of 0.2% in the anonymized subset and 0.4% for the entire cohort (Table 1). 22 patients had a germline alteration in MSH6/MSH2/MLH1/PMS2, of which 20 had Lynch Syndrome and 2 had constitutional mismatch repair deficiency (CMMRD)—a prevalence of 0.6% in the anonymized subset and 1% for the entire cohort. 8 patients were found to have a germline alteration in NF1 and have neurofibromatosis type 1—a prevalence of 0.2% in the anonymized subset and 0.4% for the entire cohort. And finally, 1 patient had a germline alteration in PTEN and has PTEN hamartoma tumor syndrome—a prevalence of 0.08% in the anonymized subset and 0.05% for the entire cohort. These 4 genetic predisposition syndromes impacted 1.8% of the patient population; no patients had a TSC1/2 mutation or an APC variant that was not I1307K.
Table 1 Germline predisposition syndromes and associated glioma/GNTTP53 mutations and MMR alterations were primarily found in 2 glioma subtypes: glioblastoma, IDH WT, and IDH mutant astrocytoma. As expected, NF1 mutations were primarily associated with IDH WT high-grade gliomas and pilocytic astrocytomas (Table 1, Fig. 2a).
Fig. 2
Germline alterations and somatic biallelic inactivation in the associated tumor. a Germline alterations by mutation and tumor type. b Breakdown of germline alteration by somatic biallelic inactivation in the associated tumor. c Age of diagnosis in patients with moderate or high penetrance germline alteration with biallelic inactivation (maroon), a monoallelic germline alteration without biallelic inactivation (pink), and no germline alteration (teal)
The remainder of the germline alterations included high and moderate penetrance genes in DNA damage response pathway alterations such as BRCA2 (n = 11; 0.5%), CHEK2 (n = 21, 0.9% excluding common lower penetrance variant I157T), and ATM (n = 8; 0.36%) (Fig. 2a).
Analysis of biallelic inactivationOur analysis revealed that nearly all patients with Li-Fraumeni and neurofibromatosis type 1 (NF1) had a second hit in their tumor (88% and 100% of tumors, respectively) (Fig. 2b, Supplemental Data Table for patient-level annotation). In patients with NF1, biallelic inactivation was identified in high and low-grade astrocytomas, but also a myxopapillary ependymoma, which is not classically associated with NF1 despite reports of co-occurrence [6, 33].
Surprisingly, in patients with Lynch syndrome and a glioma/GNT, we observed a lower rate of biallelic inactivation (10/19 patients, 53%). Among the patients with biallelic inactivation, 3/10 had high-grade IDH mutant astrocytoma and 7/10 had glioblastoma, IDH WT. Of the remaining patients without biallelic inactivation, 7/9 had glioblastoma, IDH WT; 1/9 had an IDH mutant astrocytoma; and 1/9 had a ganglioglioma.
All tumors with biallelic inactivation of a heterozygous germline mutation (n = 10) and the 2 patients with homozygous germline mutations (CMMRD) were hypermutated with a tumor mutational burden > 20 mutations/mega base. In contrast, tumors without biallelic inactivation, exclusively had a low tumor mutational burden (< 10 mutations/mega base) (Fig. 3a). Ultra-hypermutation (defined as a tumor mutational burden greater than 100 mutations/mega base) [4] exclusively occurred in tumors with a secondary oncogenic/likely oncogenic POLE mutation or occurred in the setting of CMMRD.
Fig. 3
Patients with a germline mismatch repair defect. a Tumor mutational burden in patients with Lynch without biallelic inactivation (purple), in patients with Lynch with biallelic inactivation without a secondary POLE mutation (red), in patients with Lynch with biallelic inactivation with a secondary POLE mutation (blue), and in patients with constitutional mismatch repair deficiency (green). b MSI status by the MiMSI classifier in glioma and glioneuronal tumors occurring in the setting of constitutional mismatch repair deficiency (CMMRD), biallelic inactivation of an MMR alteration and a secondary POLE mutation (Biallelic MMR + POLE), biallelic inactivation of an MMR alteration without a POLE mutation (Biallelic MMR), and a heterozygous germline MMR alteration without biallelic inactivation (Monoallelic MMR)
MSI status was determined using MiMSI on the 10 patients with Lynch syndrome or CMMRD in the un-anonymized subset. All tumors that were MSI-high (n = 8) had biallelic inactivation of an MMR gene (Fig. 3b). 2 tumors had biallelic inactivation but were microsatellite stable (MSS); these tumors had the SBS15 signature, which is indicative of defective mismatch repair, on mutational signature analysis [1] and had loss of protein expression of the impacted MMR gene on immunohistochemistry.
Biallelic inactivation was least commonly observed in Lynch syndrome patients with P/LP mutations in PMS2—only 1/6 had tumors with biallelic inactivation. In comparison, 4/7 patients with MSH2, 3/4 patients with MSH6, and 2/2 patients with MLH1 mutations had glioma/GNT with biallelic inactivation. Unlike the hypermutated glioblastoma, IDH WT tumors, which never had chromosome 7/10 alterations, EGFR amplification, or TERT promoter mutations (7 out of 7), 7 of 8 patients with an IDH WT glioma without hypermutation had these typical molecular features of glioblastoma, IDH WT. Among patients with Lynch syndrome, the median age of patients with biallelic inactivation of the germline MMR alteration was 32.4 vs a median age of 54.0 for patients with a monoallelic MMR alteration in their tumor.
Biallelic inactivation was least common in patients with germline variants that are not known to be associated with glioma/GNTs. Biallelic inactivation was found in patients with germline BRCA1/BRCA2 alterations in 21.4%, in patients with a low-penetrance germline APC I1307K germline variant in 13.7%, and in patients with MUTYH alterations in 9.5% (Fig. 2b).
Of the 14 patients with germline BRCA1 or BRCA2 alterations, a HRD score could be calculated for 10. 2/10 have tumors with biallelic inactivation of the germline alteration and the 2 highest HRD scores (31 and 49); both these tumors had BRCA2 alterations and were glioblastoma/astrocytoma, IDH WT. For the 8/10 patients with monoallelic BRCA1/BRCA2 alterations, the median HRD score was 9.5, ranging from 6 to 31 (Supplementary Table 2).
APC (specifically, I1307K) and MUTYH variants are common, accounting for 26.5% of identified variants. It has been suggested that these genes are associated with the development of glioma/GNT [2, 3, 15]. The only APC alteration identified, the APC I1307K variant, is present in ~ 6–10% of individuals of Ashkenazi Jewish ancestry and is less frequent in non-Jewish populations. This variant can contribute to colorectal cancer by causing polymerase slippage, resulting in a frameshift mutation. In tumors with this frameshift, a second somatic mutation causing biallelic inactivation is often identifiable [16, 28]. The 4 glioma/GNT identified to be biallelic for APC I1307K had loss of heterozygosity; the absence of an additional somatic mutation decreases the likelihood that this germline variant contributed to tumorigenesis. Of the 42 patients with MUTYH variants, only 4 had biallelic inactivation—1 glioblastoma, IDH WT and 3 oligodendrogliomas. The rate of biallelic inactivation in glioblastoma, IDH WT with MUTYH alteration is 3.8%; the much higher frequency of biallelic inactivation in oligodendroglioma (42.8%) is attributable to the location of MUTYH on the short arm of chromosome 1 (1p34). The 1p arm with the MUTYH mutation was lost (n = 4) or retained (n = 3) to similar extents, which suggests that biallelic inactivation fails to confer a selective advantage.
Germline alteration and clinical characteristicsPatients with a glioma/GNT and biallelic inactivation of a moderate or high penetrance germline alteration were younger (median age at diagnosis: 31.2, Fig. 2c) than the subset of patients with a germline mutation without loss of heterozygosity (median age at diagnosis: 50, p = 0.00014) and were younger than the subset without a germline alteration (median age at diagnosis: 51.4 years, p = 3.5 × 10–6).
Within the pediatric subset (age < 15 years old), 45% of patients (10/22) with germline alterations developed tumors with biallelic inactivation of an affected gene (7 tumors had biallelic inactivation of NF1, a ganglioglioma had biallelic inactivation of SUFU, and individual gliomas had biallelic inactivation of ATM and BRCA2). Compared to the pediatric subset, biallelic inactivation of the germline variant was less frequently identified in AYAs (32%, Fisher’s exact test: p = 0.20) and adults (16%, Fisher’s exact test, p = 0.0017).
Additional clinical data were available for analysis in the un-anonymized subset. Of the 5 patients with Li-Fraumeni who consented to clinical germline testing, 3 had a known germline TP53 variant, while 2 were diagnosed through this testing (a patient with and another without a history suggestive of a tumor predisposition syndrome). Unlike Li-Fraumeni, NF1 has clinical criteria that are highly sensitive and specific for this genetic syndrome. For this reason, all 6 patients with a germline NF1 alteration in the un-anonymized subset previously received a clinical diagnosis.
Among the 10 patients with an MMR-deficient tumor who underwent germline assessment, 2 patients previously tested positive for Lynch syndrome or CMMRD, 3 patients were evaluated due to a suspicious personal or family history, 1 patient underwent evaluation in the absence of clinical suspicion, and 4 patients were tested after somatic hypermutation was identified in the tumor. Of the 5 patients with Lynch syndrome with an MMR proficient tumor, 3 were screened in the absence of clinical suspicion and had low penetrance PMS2 P/LP mutations—the remaining 2 patients were previously diagnosed with Lynch Syndrome due to a personal history of cancer.
The 5 patients with Lynch syndrome with IDH WT gliomas that were MMR proficient succumbed to their tumor in a time frame that is typical for an IDH WT glioblastoma—median survival of 27 months (range of 14 to 40 months). The prognosis of the 7 patients with a germline MMR defect and a hypermutated glioma was more variable. While the median survival of the IDH WT gliomas with MMR deficiency was 30 months, 3 patients have no evidence of active disease at 52, 72, and 207 months of follow-up. Of these 3 long-term survivors, only 1 patient (the individual with the longest follow-up) has a secondary POLE mutation, which has been associated with an improved prognosis [10]. In total, 3 of 7 patients with an IDH WT hypermutated MMR-deficient tumor had a pathogenic or likely pathogenic POLE mutation and harbored the mutational signature associated with these alterations, SBS10a/b [1]. Apart from the long-term survivor, the other patients with POLE mutations succumbed to their tumor at 20 and 21 months. Among the 5 ultra-hypermutated tumors, 3 developed leptomeningeal disease (the 3 ultra-hypermutated tumors with biallelic inactivation of PMS2). None of the 3 patients with IDH mutant, MMR-deficient, high-grade gliomas have progressed after first-line treatment at 29–67 months.
Comments (0)