
Remember me




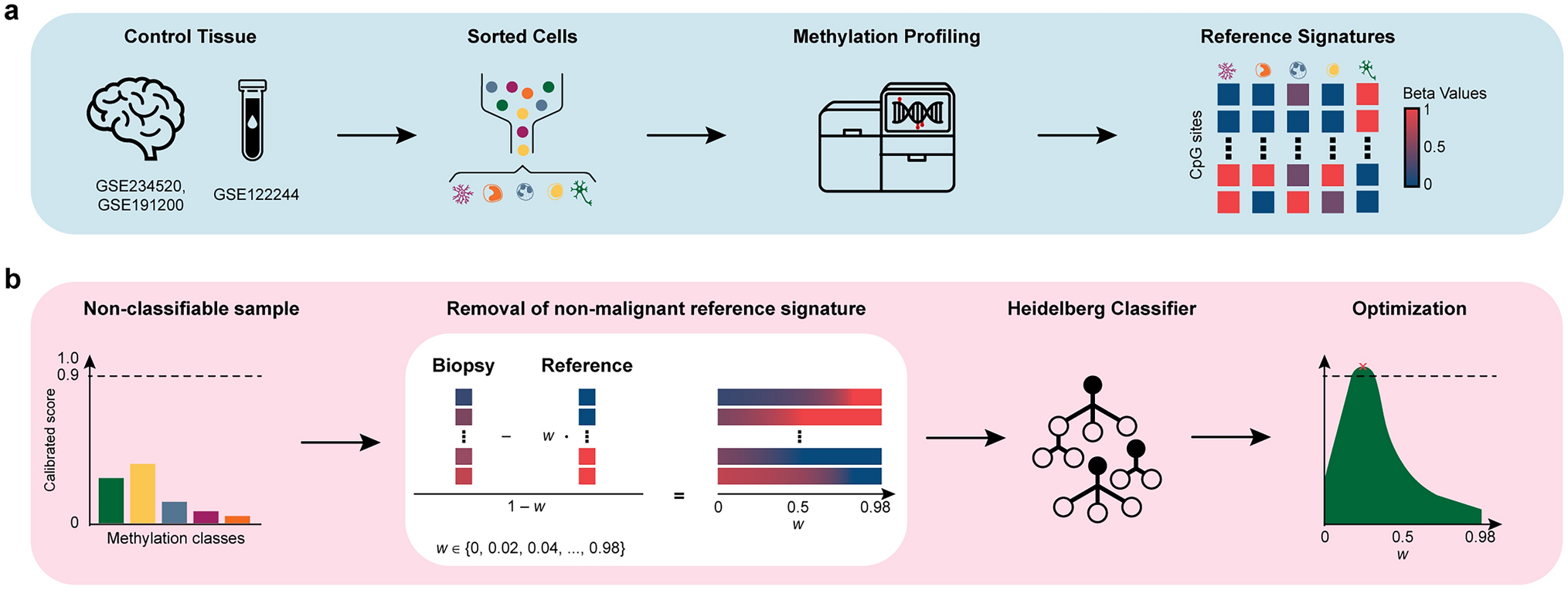


We first assessed whether our in silico purification framework improves the classification of previously non-classifiable samples by analyzing matched methylation profiles from the LOGGIC Core BioClinical Data Bank. Among 109 patients that were subjected to repeated methylation profiling, we identified 41 patients (86 samples) with at least one classifiable and one non-classifiable profile, allowing for a direct comparison between post-purification predictions and initially classifiable samples from the same patient (Table 1, Suppl. Table S3). We applied our in silico purification approach by systematically removing reference signatures of microglia, monocytes, neutrophils, T cells, and neurons. Following purification, 9 (20.5%) of the initially non-classifiable samples were confidently classifiable by the version 11b4 Heidelberg Classifier (Fig. 2). In all 9 cases the post-purification classification of the initially non-classifiable samples (first “punch”) concurred with the classification of the matched originally classifiable sample (second “punch”), indicating that our approach successfully recovered tumor-specific methylation signals from the original sampling region. To test whether these findings generalize to the updated Heidelberg Classifier version 12.8, we reanalyzed these 9 samples. Two of the nine samples were initially classifiable without purification, five became classifiable after purification, and two remained non-classifiable. Notably, all (7/7) confident predictions before and after purification were concordant with the version 11b4 results and the matched punches (Suppl. Table S4), supporting consistency across classifier versions. Moreover, while the average interval between the first and second EPIC scan dates for the 41 repeatedly profiled samples was 10.22 ± 3.95 days, the in silico purification and reclassification process, performed on an Apple M2 MacBook Air (8-core CPU, 16 GB RAM), required just 3.28 ± 0.33 s per sample.
Fig. 2
In silico purification in a validation cohort of pLGG cases subjected to repeated DNA methylation profiling. Rows represent individual cases. Columns from left to right show (1) case number, (2) initial histopathologic diagnosis, (3) methylation-based classification of the first non-classifiable punch and (4) second confidently classified punch, (5) integrated diagnosis combining histopathology with methylation-based classification, (6) results of in silico purification applied to the previously non-classifiable first punch, and (7) lists of non-malignant cell types allowing confident classification for each case. Stacked bar plots show calibrated probabilities on the y-axis, with colors indicating respective tumor classes, tumor class families, and control classes. Probabilities below 0.15 are summarized as OTHER in gray. Dashed lines indicate the 0.9 confidence threshold. Bar plots for in silico purification show predictions after removing the non-malignant cell signature that yielded the highest classification confidence, with the removed fractions shown on the x-axis (from 0 to 0.98, left to right). The corresponding cell type whose removal enabled optimal classification is highlighted in bold font
Next, we compared methylation-based predictions with both the initial histopathologic diagnosis and the integrated diagnosis, incorporating histopathology and methylation profiling, established by expert neuropathologists. In 6 of the successfully purified cases, the methylation-based prediction matched both the initial and integrated diagnoses. In all 3 remaining cases, the methylation-based classification differed from the initial histopathologic diagnosis but ultimately aligned with the integrated diagnosis, highlighting the role of methylation profiling in refining or correcting initial assessments. Specifically, these three cases were initially diagnosed as PA (case 48 and 52) and dysembryoplastic neuroepithelial tumor (DNT) (case 55), but were reclassified as rosette-forming glioneuronal tumor (RGNT), desmoplastic infantile astrocytoma/ganglioglioma (DIG/DIA) and PA, respectively, after integration of histopathology with the methylation-based predictions.
Histopathology supports purification rationaleWe observed that the removed non-malignant reference signature often corresponded to the control class for which the sample had an elevated probability. Two samples classified as hemispheric cortex control (CONTR_HEMI) became classifiable as tumor only after removal of the neuronal signature, while six samples with elevated reactive tumor microenvironment control scores (CONTR_REACT) became classifiable after removal of immune cell signatures but not the neuronal signature, suggesting that abundant non-malignant cells may mask the malignant methylation signal by contributing to control-like profiles.
To further investigate whether the initially elevated control-class scores and successful purification after removing specific non-malignant cell signatures indeed reflected a higher abundance of these cell types, we analyzed H&E-stained sections from tissue layers adjacent to the blocks used for methylation profiling in two cases (case 8 and 11), where the sampling locations of the first (non-classifiable) and second (classifiable) punches were clearly marked prior to methylation profiling. In case 11, the first punch was initially classified as CONTR_REACT, but was reclassified as PXA after removal of the T cell signature. Accordingly, the H&E section showed an increased density of lymphocytes in the region sampled for the first punch, while the second punch location showed no such lymphocyte enrichment (Fig. 3a). Similarly, in case 8, the first punch, initially classified as CONTR_HEMI and reclassified as DNT after removal of the neuronal signature, corresponded to a histologic region with a visibly greater abundance of neurons compared to the region associated with the second punch (Fig. 3b).
Fig. 3
H&E-stained sections of pLGG biopsies of case 11 (a) and case 8 (b) from the LOGGIC dataset of repeatedly profiled samples. Sections are directly adjacent to the tissue used for methylation profiling. Dashed ovals indicate locations of the first and second punches. Magnifications on the left show representative areas from each punch site
Over 20% of non-classifiable samples become classifiable after purificationTo assess the impact of our in silico purification approach on a larger cohort, we collected methylation array data of pLGGs from Sturm et al. (n = 410; Table 1, Suppl. Table S5). Among all samples, 302 (73.7%) were classifiable using version 11b4 of the Heidelberg Classifier and therefore outside the intended scope of the purification algorithm. To assess whether purification could inadvertently change classifications, we applied the algorithm to these 302 cases. While 72.2% (n = 218) of them achieved slightly higher scores in their originally predicted tumor class after removal of non-malignant signatures, none of them changed their highest scoring classification (Fig. 4a).
Fig. 4
In silico purification applied to an external pLGG dataset. a Scatter plot of all 410 pLGG samples from the Sturm dataset comparing the probability scores for the highest-scoring tumor class (or tumor class family when applicable) before (x-axis) and after (y-axis) purification. Black lines mark the 0.9 classification threshold. Dots are colored by class changes, with green for stable classification, yellow for tumor class changes, and grey for initial classification as control. b Sankey plot comparing the initial highest-scoring class with the confidently predicted class after purification for all initially non-classifiable pLGG samples from the Sturm dataset that became classifiable through purification (n = 26). c Matrix comparing histopathological diagnoses with classifications after purification for the 26 pLGG samples from the Sturm dataset that became classifiable through purification. Numbers indicate sample counts. Purple represents concordance or refinement, and orange indicates discrepancy
The remaining 108 samples (26.3%) were non-classifiable, either scoring highest for a control class (n = 34) or failing to reach the confidence threshold of 0.9 (n = 74), highlighting the challenge of low classification rates in pLGGs. Our in silico purification approach enabled classification in 26 (24.1%) samples (Fig. 4a). Notably, 13 of 34 samples (38.2%) initially scoring highest in a control class became classifiable, indicating that especially samples initially classifying as non-malignant control tissues benefit from purification. In contrast, 13 of 74 samples (17.6%) initially scoring highest in a tumor class became classifiable. Of these, all 10 with an initial highest tumor class score between 0.5 and 0.9 retained their original highest scoring tumor class, while 2 of the 3 samples with an initial highest score below 0.5 changed classification, both shifting from GBM to DIG/DIA (Fig. 4b). Post-purification prediction matched or refined the initial histopathologic diagnosis in 20 of the 26 newly classifiable cases (76.9%). In the remaining 6 cases, it differed from the initial diagnosis (Fig. 4c). In all cases with available DNA or RNA sequencing data, the results were compatible with the post-purification classification (Suppl. Table S5).
We extended our analysis to a second validation dataset published by Hardin et al. consisting of 113 pLGG samples (Table 1, Suppl. Table S6). Consistent with the results from the Sturm cohort, all 91 initially classifiable samples in the Hardin dataset retained their original predicted tumor class after purification (Fig. S1a). Among the 22 initially non-classifiable samples, 5 (22.7%) became classifiable after purification. Of these, 3 transitioned from control to tumor classes, while the remaining 2, initially predicted as DNT with low confidence, reached the threshold for a confident classification post-purification (Fig. S1b). In all 5 cases, the final methylation-based classification either confirmed or refined the histopathologic diagnosis (Fig. S1c).
We further evaluated our framework using the updated version 12.8 of the Heidelberg Classifier (Suppl. Table S5). Applied to the Sturm et al. cohort (n = 410), we observed that the initial confident classification rate was higher with version 12.8 than with 11b4 (78.5% vs. 73.7%). However, 88 samples (21.5%) remained unclassifiable. Applying our purification framework to these samples enabled confident classification for 31 cases (35.2%). Of these, 21 also had a confident pre- or post-purification prediction using the version 11b4 classifier. Classifications were concordant in 18 of these cases (85.7%). Among the 10 samples that became classifiable only after purification with version 12.8 but remained non-classifiable with version 11b4, four were concordant with histopathology, while six showed discordant predictions, including classifications outside the low-grade glioma spectrum. Notably, four of these cases required removal of more than 30% of the reference signature to reach optimal classification scores. Thus, while predictions that only emerge after extensive removal of reference signatures should be interpreted with caution, the vast majority of version 12.8 post-purification classifications are consistent with version 11b4 or histopathology.
Purification refines methylation profiles for samples of moderate purityWhile our algorithm reports results for the reference cell type that maximizes tumor class probabilities, we investigated whether samples could become classifiable by removing different cell types or only one specific cell type. For the Sturm et al. cohort, the highest number of samples became classifiable upon neutrophil or monocyte removal (n = 23), with 22 responding to both and only 2 exclusively dependent on one (Fig. 5a). Microglia removal enabled classification in 19 and T cell removal in 14 samples, nearly all overlapping with samples classifiable by monocyte and neutrophil removal. Neuronal signature removal had the least impact, classifying only 5 samples, 4 of which were already classifiable through other cell type removals, indicating a high abundance of multiple non-malignant cell types in these samples. Similar results were obtained for the Hardin et al. cohort, in which four of five samples became classifiable after removing immune cell signatures from microglia, monocytes, neutrophils, or T cells, suggesting a higher degree of similarity among these cell types, while the remaining sample required neuronal signature removal for classification (Fig. S1d).
Fig. 5
Refinement of DNA methylation profiles using in silico purification. a Matrix showing the 26 pLGG samples from the Sturm dataset that became classifiable through purification, indicating how many became classifiable by removing each of the five cell types and the overlap between samples classifiable under either removal in each cell type pair. Numbers and color intensity represent sample count. b Histograms showing the fraction of non-malignant cell signal removed for optimal classification in all 410 pLGG samples from the Sturm dataset. Samples are grouped by classification status: initially classifiable (left), newly classifiable post-purification (middle), and non-classifiable (right). c t-SNEs of 579 Capper reference samples, comprising control (n = 68) and low-grade glioma (n = 511) cases, combined with either the unpurified (left) or purified (right) methylation profiles of the 26 Sturm samples that became classifiable after purification. Colors indicate the highest-scoring methylation class. The 26 Sturm samples are shown as larger, numbered dots to allow for comparison across panels
Interestingly, when the algorithm was applied to initially classifiable samples of the Sturm et al. cohort, only small fractions of non-malignant signatures were removed, typically ≤ 10% (93%, n = 281) and never exceeding 25%, suggesting that purification had little impact on strong initial classifications (Fig. 5b). In contrast, samples that became classifiable after purification required 10 to 50% subtraction to maximize tumor class scores, suggesting that moderate abundance of non-malignant cells obscured tumor signals but could be computationally corrected. Non-classifiable cases that remained unclassified post-purification showed a bimodal pattern: 51.2% (n = 42) reached their maximum score with < 20% removal, and 34.1% (n = 28) with ≥ 60%. Similar observations were made for the Hardin et al. cohort (Fig. S1e).
To further assess the impact of purification on methylation profiles, we generated paired t-SNE plots of either the unpurified or optimally purified profiles of the 26 Sturm et al. samples that became classifiable following purification, combined with reference low-grade glioma (n = 511) and control (n = 68) samples. The embedding based on the purified profiles appeared visibly sharpened and showed clear repositioning: 7 samples (26.9%) shifted from control clusters toward their corresponding tumor clusters, while 2 additional samples showed directional movement toward their target clusters without fully reaching them (Fig. 5c). Another 7 samples that were already positioned close to their tumor clusters moved post-purification even closer to their cluster centers, and 6 samples maintained their appropriate initial positioning. Overall, 16 of the 26 samples (61.5%) showed improved spatial positioning following purification, and 6 additional samples maintained favorable locations, indicating that purification not only enables confident classification using the Heidelberg Classifier but also sharpens underlying tumor methylation profiles.
Lastly, to investigate why many initially non-classifiable samples remained unclassifiable despite purification, we examined array-level quality control metrics across initially classifiable, newly classifiable, and persistently non-classifiable samples. In all three datasets (LOGGIC, Sturm, and Hardin), all samples with more than 1% of probes showing detection p-values greater than 0.01 were persistently non-classifiable (Fig. S2, Suppl. Table S7), suggesting that in some cases, technical quality issues rather than low tumor cell content limit classifiability.
Comments (0)