
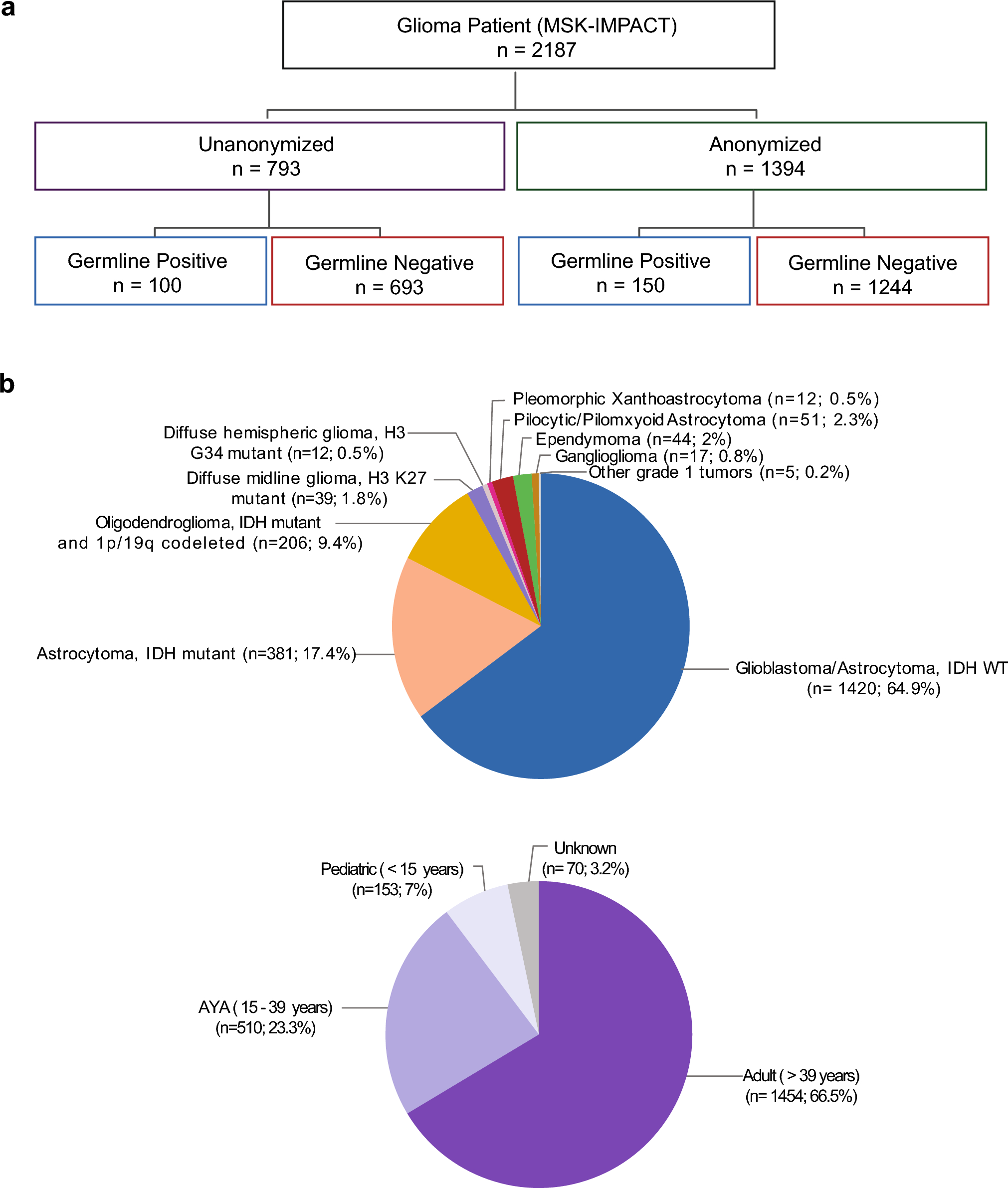
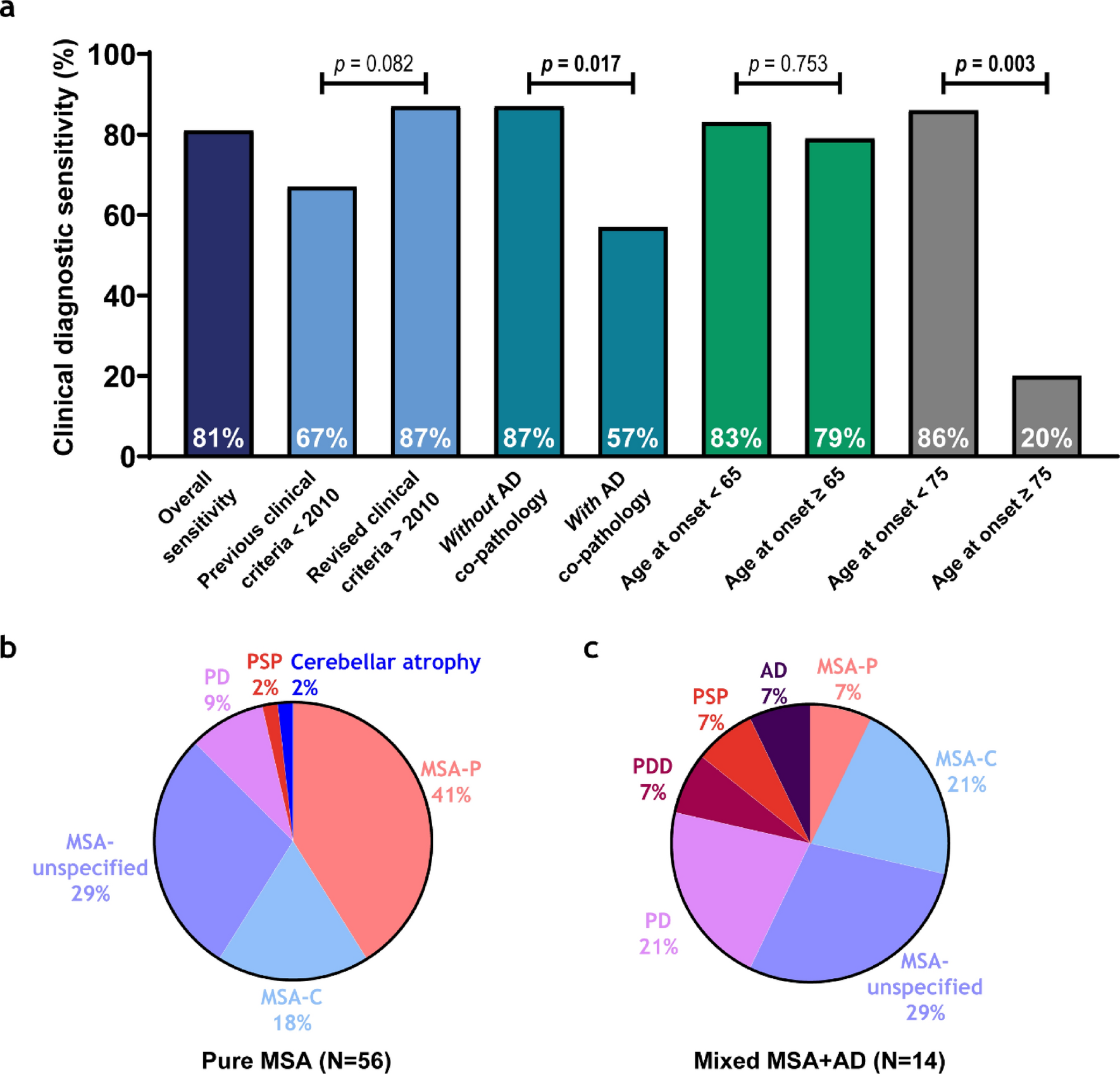
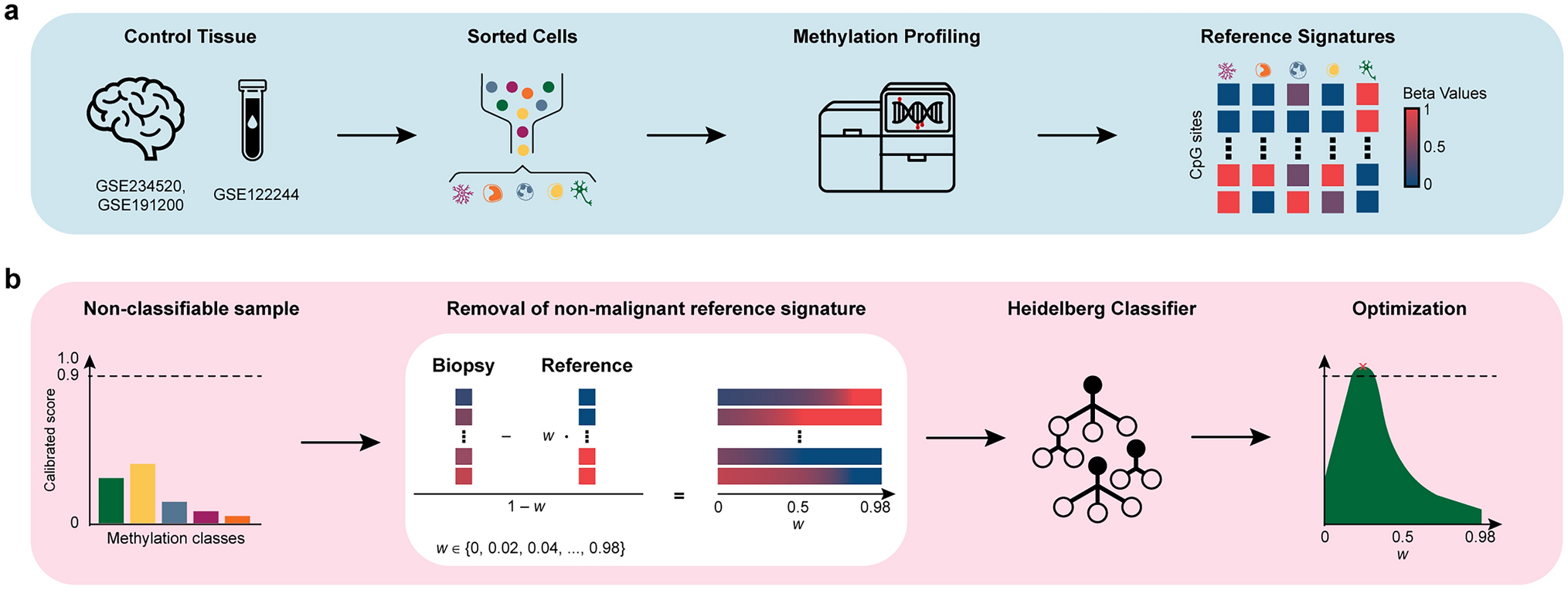
Remember me






Middle temporal cortex samples of pathologically confirmed AD cases were obtained from the Mayo Clinic Brain Bank for neurodegenerative diseases. At the outset of this study, the focus was to investigate the impact of APOE genotypes on lipid signatures in the cerebrovasculature. To this end, we initially enrolled 23 cases carrying the APOE ε2 allele (ε2/ε3, N = 9; ε2/ε4, N = 14). Subsequently, we enrolled age-, sex-, CAA-score matched cases from the APOE ε3/ε3 (N = 21), ε3/ε4 (N = 23), and ε4/ε4 (N = 22) genotypes. Participants were required to meet inclusion criteria: (1) age greater than 60 years, (2) Thal phase III or higher [62], and (3) Braak stage IV or higher [7]. Individuals treated with antibody-based AD immunotherapies against Aβ or tau were not included. This resulted in a total of 89 cases, all of whom were non-Hispanic and White. We also collected demographic data (age of death and sex) as well as pathological information, including average CAA scores, Braak stage, Thal phase, and arteriolosclerosis pathology grading. To represent the diverse spectrum of an autopsied AD population, cases with comorbid neuropathologies were included in this study (Supplementary Table 1). The demographic and pathological characteristics of the cohort are summarized in Table 1. No significant differences in age, sex, pathological scores, or cerebrovascular Aβ and tau levels were observed across APOE genotypes. Genomic DNA was extracted from frozen brain tissue using the standard protocols. The APOE single nucleotide variants (rs429358 C/T and rs7412 C/T), which define the APOE ε2, ε3, and ε4 alleles, were genotyped using custom TaqMan Allelic Discrimination Assays on a QuantStudio 7 Flex Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). Genotype calls were generated using TaqMan Genotyper Software v1.3 (Applied Bio-Systems). Genotype call rates were 100% for each variant, and no deviations from the Hardy–Weinberg equilibrium were observed (all p > 0.01). Experimental procedures were conducted in compliance with protocols approved by the Mayo Clinic Institutional Review Board.
Table 1 Summary of participant characteristics overall and separately according to APOE groupHistopathologic assessment for cerebrovascular diseaseAll cases were subjected to standardized neuropathological sampling and evaluation as previously described [56]. We assessed AD neuropathologic changes, including Braak tangle stage and Thal amyloid phase, as described previously [7, 46, 62]. CAA staging of parenchymal vessels was performed using the method previously described [57]. Thioflavin-S staining was used to evaluate the severity of CAA and was scored in 5 cortical regions: superior temporal cortex, inferior parietal cortex, middle frontal cortex, motor cortex, and visual cortex. A semi-quantitative approach was employed with the following scoring system: 0, the absence of amyloid-positive vessels; 0.5, amyloid deposition limited to the leptomeninges; 1, mild amyloid deposition in both leptomeninges and parenchymal vessels; 2, moderate circumferential amyloid deposition in some vessels; 3, widespread severe amyloid deposition in leptomeninges and parenchymal vessels; 4, reflecting more severe CAA with dysphoric changes (Fig. 1a-d). The average CAA scores were defined as the mean of region-specific CAA scores from five cortical regions. Hematoxylin and eosin staining sections obtained from superior temporal cortex were assessed for arteriolosclerosis according to the vascular cognitive impairment neuropathology guidelines (VCING) [16, 58]. Severities were scored using a semi-quantitative method as follows: Arteriolosclerosis: 0, normal; 1, mild fibrosis with mild medial thickening; 2, moderate fibrosis; 3, severe fibrosis, based on overall impression (Fig. 1e-g).
Fig. 1
Morphologies of arteriolosclerosis and Cerebral amyloid angiopathy (CAA) with different scores in AD brains. CAA score was assessed using thioflavin-S fluorescence (green). a Sparse amyloid presence in both leptomeningeal and cortical vessels was rated as score 1. b Strong, circumferential amyloid deposition in some, but not all vessels were rated as 2. c Widespread, strong amyloid deposition in both leptomeningeal and cortical vessels were rated as score 3. d The most severe cases, exceeding the criteria for score 3 were rated as 4. Arteriolosclerosis score was assessed using hematoxylin and eosin-staining. e Cases with mild arteriolosclerosis, characterized by mild fibrosis and slight medial thickening were rated as 1. f Cases with moderate arteriolosclerosis, with evident fibrosis and moderate medial thickening were rated as 2. g Cases with severe arteriolosclerosis, marked by extensive fibrosis and pronounced medial thickening were rated as 3. Scale bars: 200 µm (A, B); 100 µm (C, D); 40 µm (E); 50 µm (F, G)
Isolation of human cerebrovasculaturesWe isolated vessels from middle temporal cortex samples dissected from AD patients, following a previously described protocol with minor modification [6] (Supplementary Fig. 1a). All procedures were conducted on ice. After the removal of meninges with tweezers, we thawed each temporal cortex sample (400 mg) on ice in vascular isolation buffer (10 μM HEPES (Thermo Fisher Scientific, Cat No. 15630080, Waltham, MA, USA) in HBSS (Thermo Fisher Scientific, Cat No. 14025092)). We manually minced the brains with a scalpel in the vascular isolation buffer to obtain small pieces of brain tissues and transferred them to a 7 ml tissue grinder. We homogenized the samples, transferred to 15 ml conical tubes, and centrifuged at 1000 g for 10 min at 4 °C. We removed the supernatants, suspended the pellet with 5 ml of vascular isolation buffer containing 18% dextran (from leuconostoc mesenteroides, M.W. 70,000; Sigma-Aldrich, Cat No. 31390, St. Louis, MO, USA), and centrifuged at 4000 g for 20 min at 4 °C. We then discarded the myelin layer with the supernatant, carefully wiped the inside wall of the conical tube with absorbent paper and resuspended the residual pellet with 1 ml of vascular isolation buffer. Next, we sequentially filtered the resuspended homogenates using 100 μm and 20 μm nylon filters in succession (Pluriselect, Cat No. 43–50,100-51 and 43–50,020-03, Leipzig, Germany). We defined the homogenates retained on the 100 μm filter as large vessel fractions, comprising large-sized vessels, such as leptomeninges, penetrating arteries and arterioles. Homogenates retained on the 20 μm filter were defined as small vessel fractions, primarily consisting of capillaries. We also defined the filtrates mainly containing vascular-depleted parenchymal tissues as parenchymal fractions. Each retained vascular fraction was washed off from filters with 500 μl of lysis buffer (150 mM NaCl, 10 mM NaH2PO4, 1% Triton X-100 (Sigma-Aldrich, Cat No. T9284), 0.5% SDS (Invitrogen, Cat No. AM9820, Carlsbad, CA, USA) and 0.5% sodium deoxycholate (Thermo Fisher Scientific, Cat No. 89904) containing phosphatase inhibitors (Roche Diagnostics, Cat No. 4906845001, Indianapolis, IN, USA) and protease inhibitors (Roche Diagnostics, Cat No. 11697498001), and 1 mM EDTA. Each fraction was placed on ice for 30 min, sonicated, and centrifuged at 100,000 g for 20 min at 4 °C in an ultracentrifuge tube (Beckman Coulter, Cat No. 357448, Indianapolis, IN, USA). The vascular-depleted parenchymal tissues were centrifuged at 16,000 g for 20 min at 4 °C, and pellets were homogenized in 500 μl of lysis buffer, sonicated and centrifuged at 100,000 g for 20 min at 4 °C. The supernatants derived from each fraction were collected and stored at −80 °C (referred to as soluble fraction). We further homogenized the pellets with 125 μl of formic acid (Sigma-Aldrich, Cat No. 33015), sonicated, and centrifuged at 16,000 g for 20 min at 4 °C. After evaporating formic acid, the pellets were resuspended in 350 μl of 5 M guanidium solution in Tris–HCl 50 mM and stored at -80 °C (referred to as the insoluble fraction). We determined protein concentrations of all fractions using the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Cat No. 23225). We also isolated vessels for lipidomic analyses using the same set of AD brain samples as separate single filtration step. The resuspended vascular homogenates were filtered through a 20 μm nylon filter (Pluriselect, Cat No. 43–50,020-03). The retained vascular fraction was washed off from the filter with 500 μl of vascular isolation buffer, sonicated, and centrifuged at 100,000 g for 20 min at 4 °C in ultracentrifuge tubes. The supernatant was aspirated, and the pellet was stored at −80 °C for subsequent lipidomic analysis.
Preparation of homogenized whole brain tissuesBrain tissue from the middle temporal cortex (50 mg) was mechanically homogenized in lysis buffer (150 mM NaCl, 10 mM NaH2PO4, 1% Triton X-100, 0.5% SDS, and 0.5% sodium deoxycholate) containing phosphatase and protease inhibitors. The brain homogenates were subsequently ultracentrifuged at 100,000 × g for 1 h at 4 °C. The supernatant was then collected as whole brain fractions (W) and stored at −80 °C for western blot analysis to compare with the vascular-enriched and vascular-depleted parenchymal fractions.
Enzyme-linked immunosorbent assay for Aβ and tauWe determined Aβ40 and Aβ42 levels of vascular-enriched fractions using highly sensitive ELISA kits (Fujifilm Wako Pure Chemical Corporation, Cat No. 292–62,301 and 296–6440, Osaka, Japan) according to the manufacturer’s instructions, as previously described [63]. We also determined phospho-tau (Thr231) and total tau levels using a kit (Meso Scale Diagnostics, Cat No. K15121D, Rockville, MD, USA) according to the manufacturer’s instructions. Signal detection was performed using MESO QuickPlex SQ 120MM and data analysis was carried out with Methodical Mind software (Meso Scale Diagnostics). All measurements were normalized against total protein amounts.
LipidomicsLipid species were analyzed using multidimensional mass spectrometry-based shotgun lipidomic analysis [25]. In brief, each sample homogenate containing 0.5 mg of protein which was determined with a Pierce BCA assay was accurately transferred to a disposable glass culture test tube. A pre-mixture of lipid internal standards (IS) was added prior to lipid extraction for quantification of the targeted lipid species. Lipid extraction was performed using a modified Bligh and Dyer procedure [67], and each lipid extract was reconstituted in chloroform: methanol (1:1, v:v) at a volume of 400 µL/mg protein. For shotgun lipidomics, lipid extract was further diluted to a final concentration of ~ 500 fmol total lipids per µL. Mass spectrometric analysis was performed on a triple quadrupole mass spectrometer (TSQ Altis, Thermo Fisher Scientific) and a Q Exactive mass spectrometer (Thermo Scientific, San Jose, CA), both of which were equipped with an automated nanospray device (TriVersa NanoMate, Advion Bioscience Ltd., Ithaca, NY) as described [28]. Identification and quantification of lipid species were performed using an automated software program [68, 73]. Data processing (e.g., ion peak selection, baseline correction, data transfer, peak intensity comparison, and quantitation) was performed as described [73]. The results were normalized to protein content (nmol lipid/mg protein).
Weighted correlation network analysisWeighted gene correlation network analysis (WGCNA) [38] was performed using quantile-normalized and log2-transformed lipid abundance measurements. Based on the relationship between power and scale independence, a power of 6 was chosen to build scale-free topology using signed hybrid network. We set the minimum modules size as 5, and merged modules whose correlation coefficients were greater than 0.7 (mergeCutHeight = 0.3). Lipid modules were represented in colors, and the eigengene (ME) of each module was tested for correlation with a series of genetic and clinical traits, including age, sex (male was coded as 1 and female was coded as 0), APOE ε2 allelic numbers, APOE ε4 allelic numbers, Braak stage, Thal phase, average CAA score, arteriosclerosis score, Aβ40, Aβ42, total tau, and p-tau231 values. Modules correlated with these traits were visualized using VisANT [33] and annotated using lipid ontology (LION) enrichment analysis [45].
Assessment of acyl chain compositions in lipidsLipid molecular species levels were converted to lipid sum composition level using LIPID MAPS, defined by chain length (total number of carbon atoms) and double bonds (DB). Abundances at the sum composition level within each lipid class were aggregated by chain length and DB number. (Supplementary material). Spearman rank correlations were then calculated between the aggregated abundances (grouped by the carbon number and DB number), APOE genotype, and cerebrovascular Aβ and tau levels. Heatmaps showed the correlations between chain length and DB number of lipid species with colors representing the correlation coefficients.
Immunostaining of isolated cerebrovasculatureThe vascular-enriched pellets were resuspended in 100 μl of 1% bovine serum albumin (BSA) (Roche, Cat No. 10735078001, Mannheim, Germany) in PBS. In parallel, the vascular-depleted parenchymal fraction was resuspended in 200 μl of 1% BSA in PBS. Vascular-enriched and vascular-depleted parenchymal fractions were deposited on glass slides (10 μl/ per slide) and left at room temperature for 30 min. Samples were fixed with ice-cold methanol, then permeabilized with PBS containing 0.3% Triton X-100 (Sigma-Aldrich, Cat No. T9284) and blocked with Protein Block Serum-Free Ready-To-Use (Agilent Technologies, Cat No. X0909, Santa Clara, CA, USA). They were then incubated overnight at 4 °C with anti-alpha smooth muscle actin (α-SMA) antibody (1:100 dilution; Abcam, Cat No. ab5694, Cambridge, MA, USA), anti-platelet-derived growth factor receptor β (PDGFRβ) (1:100 dilution; Abcam, Cat No. ab32570), anti-Type IV collagen (1:500 dilution; MilliporeSigma, Cat No. AB769, Burlington, MA, USA), and anti-NeuN (1:100 dilution; Abcam, Cat No. ab177487) diluted in background-reducing dilution buffer (Agilent Technologies, Cat No. S3022), subsequently incubated with Alexa Fluor 488 conjugated donkey anti-rabbit secondary antibody and Alexa Fluor 594 conjugated donkey anti-goat secondary antibody (1:500 dilution; Invitrogen, Cat No. A21206 and A11058) for 2 h at room temperature. The samples were mounted with DAPI hard set mounting medium (Vector Laboratories, Cat No. H1500, Burlingame, CA, USA). The images were captured using a Keyence BZ-X800 fluorescence microscope (Keyence, Osaka, Japan).
Western blottingThe soluble fractions derived from vascular-enriched, vascular-depleted parenchymal fractions, and whole brain fractions were mixed with Laemmli’s loading sample buffer and heated 5 min at 95 °C. Equal amounts of proteins per sample (10 μg) were electrophoresed on 4–20% Tris–glycine sodium dodecyl sulfate–polyacrylamide gels (Bio-Rad, Cat No. 5671094, Hercules, CA, USA). Proteins were electroblotted on polyvinylidene fluoride (PVDF) membranes (MilliporeSigma, Cat No. IPVH00010), which were then blocked for 2 h at room temperature with a PBS containing 5% non-fat dry milk. The membranes were incubated overnight at 4 °C with the primary antibodies: anti-occludin (1:1000 dilution; Invitrogen, Cat No. 71–1500), anti-α-SMA antibody (1:1000 dilution; Abcam, Cat No. ab5694), anti-PDGFRβ (1:2000 dilution; Abcam, Cat No. ab32570), anti-NeuN (1:1000 dilution; Abcam, Cat No. ab177487). The membrane was then probed with horseradish peroxidase (HRP)-conjugated secondary antibodies (Abcam, Cat No. ab6721 and ab6789). The blots were detected using Pierce™ Western Blot Signal Enhancer (Thermo Fisher Scientific, Cat No. 21050) and the ChemiDoc Touch Imaging System (Bio-Rad).
Generation of human iPSC-derived vascular mural-like cellsWe used human induced pluripotent stem cell (iPSC) lines from the male-derived KOLF2.1 J line obtained from the iPSC Neurodegenerative Disease Initiative (iNDI), consisting of isogenic iPSC lines with APOE ε3/ε3 or ε4/ε4 [52]. The characterization of the parental KOLF2.1 J line has been reported previously [52]. Human iPSC-derived vascular mural-like cells (iVMLCs) were differentiated as previously described with modifications [19]. Briefly, iPSCs were dissociated into single cells using Accutase (Stem Cell Technologies, Cat No. 07920, Vancouver, Canada) and reseeded at 25,000 cells /cm2 on Matrigel-coated plates in mTeSR1 medium supplemented with 10 μM Y27632 (Stem Cell Technologies, Cat No. 72302) for the first 24 h. To initiate the differentiation, medium was changed to STEMdiff™ Mesoderm Induction Medium (Stem Cell Technologies, Cat No. 05221). The medium was exchanged every 24 h. On day 6, cells were dissociated using Accutase and re-seeded at 35,000 cells/ cm on Matrigel-coated wells in pericyte medium (ScienCell Research Laboratories, Cat No. 1201, Carlsbad, CA, USA) and cultured for additional 8 days. All cultured cells were maintained at 37 °C in a humidified incubator with 5% CO₂.
Immunostaining of iPSC-derived vascular mural-like cellsFor immunostaining, human iVMLCs were fixed with 4% paraformaldehyde for 15 min and subsequently washed three times with PBS. Following fixation, cells were permeabilized using PBS containing 0.3% Triton X-100 (Sigma-Aldrich, Cat No. T9284) and blocked with Protein Block Serum-Free Ready-To-Use solution (Agilent Technologies, Cat No. X0909). Primary antibody incubation was performed overnight at 4 °C in a background-reducing dilution buffer (Agilent Technologies, Cat No. S3022) using mouse monoclonal anti-αSMA antibodies (Sigma-Aldrich, Cat No. A2547). After washing, cells were incubated with Alexa Fluor 488 conjugated donkey anti-mouse secondary antibody (Invitrogen, Cat No. A21202) for 2 h at room temperature. Nuclei were visualized by counterstaining with DAPI (Thermo Fisher Scientific, Cat. 62,248) at a 1:5000 dilution in PBS. Fluorescence images were acquired using a Keyence BZ-X800 microscope (Keyence, Osaka, Japan).
RNA isolation and real-time PCR analysisFor comparative gene expression studies, we extracted total RNA from human iVMLCs, human primary brain vascular smooth muscle cells (HBSMC; ScienCell Research Laboratories, Cat. No. 1100), and immortalized human cerebral microvascular endothelial cells, clone D3 (hCMEC/D3; MilliporeSigma, Cat. No. SCC066) using the RNeasy Mini Kit (QIAGEN, Cat No. 74104, Valencia, CA, USA) according to the manufacturer’s instructions. Reverse transcription was performed with the iScript™ cDNA Synthesis Kit (Bio-Rad, Cat No. 1708890) to synthesize complementary DNA (cDNA) from total RNA. For quantitative real-time PCR analysis, cDNA samples were combined with gene-specific primers and SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad, Cat No. 1725271). Amplification and detection were performed using the QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Data were processed using the ΔΔCT method with QuantStudio 7 Flex system software. Gene expression was assessed using predesigned primer sets (Integrated DNA Technologies, Coralville, CA, USA) for ACTA2 (Hs.PT.56a.2542642), PDGFRB (Hs.PT.58.22892761), OCLN (Hs.PT.58.15235048), PECAM1 (Hs.PT.58.19487865), and ACTB (Hs.PT.39a.22214847). The relative gene expression was normalized to ACTB.
Cell culture and Aβ treatmentLyophilized Aβ40 and Aβ42 peptides (Anaspec, Cat No. AS-24235 and AS-20276, Fremont, CA, USA) were dissolved in dimethyl sulfoxide (DMSO) (BioWorld, Cat No. 40470005, Dublin, OH, USA) and aliquoted for storage at −80 °C as stock solutions. On day 8, medium was replaced with non-FBS containing pericyte medium, and then human iVMLCs were treated with Aβ40 or Aβ42 for 48-h incubation. DMSO diluted in PBS (0.2% DMSO, corresponding to the amount contained in Aβ) was used for vehicle control. After treatment, cells were rinsed with PBS and were subsequently lysed using Mammalian Cell Lysis Buffer 5X (Abcam, Cat No. ab179835) supplemented with phosphatase inhibitors and protease inhibitors. The lysates were centrifuged at 1500 rpm for 5 min at 4 °C. The resulting supernatants were collected and stored at −80 °C until further analysis.
Assay of sphingomyelinase activity and quantification of sphingolipidsNeutral sphingomyelinase (nSMase) activity was measured using the Sphingomyelinase Assay Kit (Abcam, Cat No. ab138876). Quantitative analyses of sphingomyelin and ceramide were performed with the Sphingomyelin Assay Kit (Abcam, Cat No. ab133118) and Human Ceramide Elisa Kit (AFG Scientific, Cat No. EK710698, IL, USA), following the manufacturers’ instructions.
MTT assayCell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Roche Diagnostics, Cat No. 11465007001) following the manufacturer’s instructions. After 48 h of Aβ treatment, 10 μL of MTT reagent were added to each well and incubated for 4 h at 37 °C. Then, 100 μL of solubilization buffer was added, and the plates were incubated overnight at 37 °C. Absorbance was then measured at 550 and 600 nm using a microplate reader, and the difference between the two wavelength values was used to determine cell viability.
BrdU assayCell proliferation was evaluated using 5-bromo-2′-deoxy-uridine (BrdU) colorimetric assay (Abcam, Cat No. ab126556) following the manufacturer’s instructions. After 48 h of Aβ treatment, BrdU was added for 18 h, followed by incubation with anti-BrdU antibody and peroxidase-conjugated secondary antibody. The reaction was visualized using TMB substrate, stopped with stop solution, and absorbance was measured at 450 nm using a microplate reader.
Statistical analysisContinuous variables were summarized with the sample median and range. Categorical variables were summarized with the number and percentage of subjects. Comparisons of characteristics according to APOE group were conducted using a Kruskal–Wallis rank sum test (continuous and ordinal variables) or Fisher’s exact test (categorical variables). Due to distributional skewness, in the subsequently described regression analyses, a base-2 logarithm transformation was utilized for AD-related molecules including cerebrovascular Aβ40, Aβ42, t-tau, and p-tau231. A square root transformation was utilized for average CAA score, and a rank transformation was used for lipid analytes and lipid class sums. Associations of average CAA score, arteriolosclerosis score, Thal phase, Braak stage, age, sex, presence of the APOE ε2 allele, number of APOE ε4 alleles, and AD-related molecules with lipid analytes and lipid class sums were examined using unadjusted and multivariable linear regression models. Regression coefficients (denoted as β) and 95% CIs were estimated and were interpreted as the change in the mean lipid analyte or lipid class sum rank corresponding to a specified increase (continuous variables) or presence of the given characteristic (categorical variables). Multivariable models were adjusted for age, sex, number of APOE ε4 alleles (except in analysis involving presence of the APOE ε2 allele), Braak stage, and Thal phase. We utilized a Bonferroni correction for multiple testing separately for analysis assessing associations with lipid class sums (10 tests for each characteristic, p < 0.005 considered as significant) and for analysis examining associations with lipid analytes (122 tests for each characteristic, p < 0.00041 considered as significant). All statistical tests were two-sided. Statistical analyses were performed using SAS (version 9.4; SAS Institute, Inc., Cary, North Carolina). Results through iVMLCs were analyzed by one-way ANOVA followed by Tukey’s post hoc test using GraphPad Prism (Version 10; GraphPad Software Inc., San Diego, CA, USA) and p < 0.05 was considered as significant. Lipid acyl chain compositions were analyzed in R (v4.4.0). Spearman rank correlation analyses were performed by the rstatix package (v0.7.2), and heatmaps were visualized using the ComplexHeatmap package (v2.20.0) [23].
Comments (0)