
Remember me

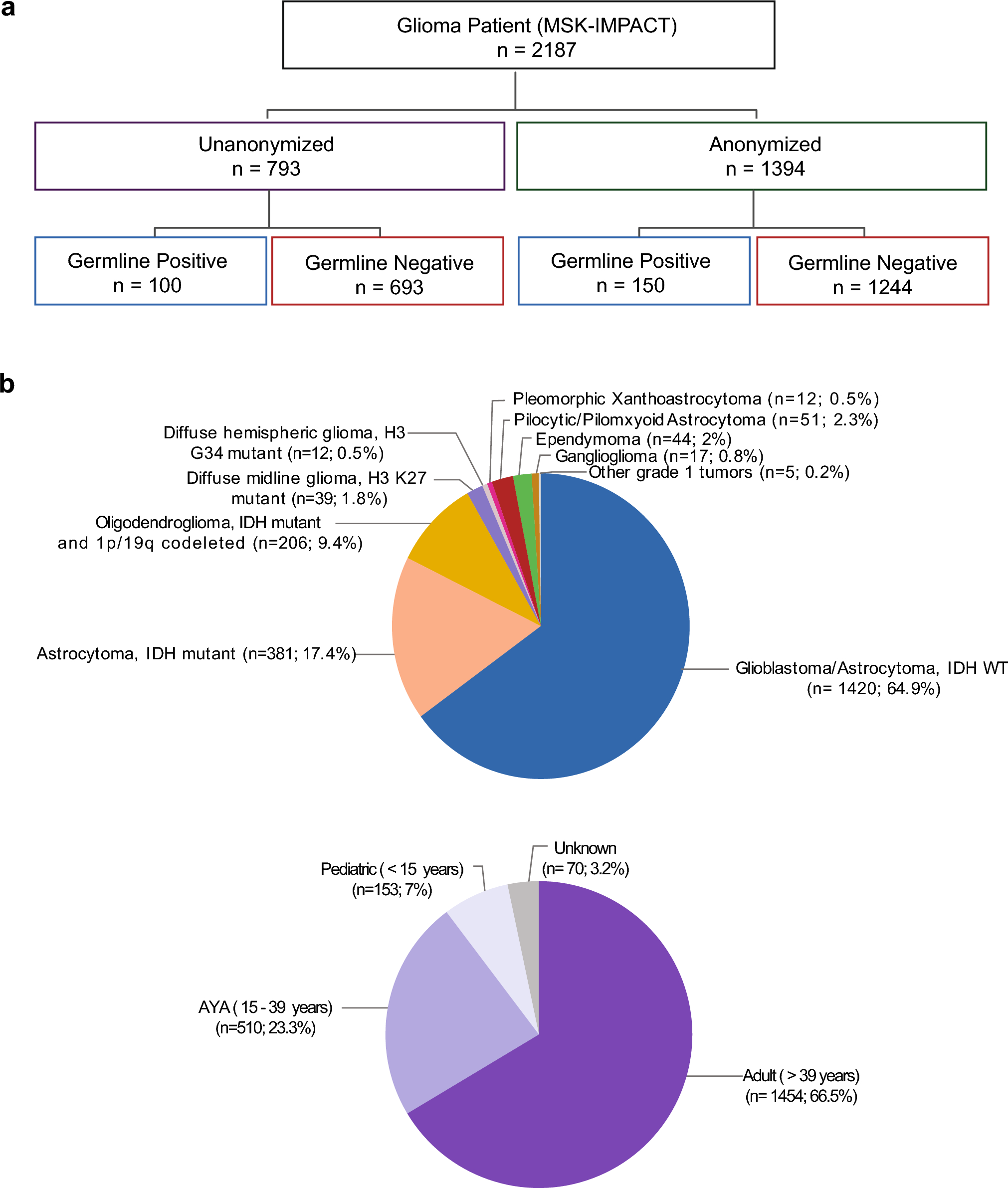





Since the initiation of the INFORM registry in 01/2015 until the data cut-off for the current study 11/2023, a total of about 3000 patients spanning all entities have been enrolled. Based on the above-mentioned selection criteria, 162 patients belonging to the DMG K27-altered subgroup were included in the current study (Supplementary Fig. 1). The patients were enrolled by 33 German pediatric oncology centers, as well as by centers in Austria, Belgium, the Netherlands, Switzerland, Sweden, Norway, Finland, Poland, the Czech Republic, Greece and Israel.
Median patient age at the episode of the disease leading to enrollment in INFORM was 8 years, with 12.3% of patients being 4 years of age or less. Pons/brainstem was the most frequent tumor localization (60.5% of patients), followed by the thalamic region (11.7%). Around three-quarters of patients (75.9%) had localized disease at the current episode. Most patients (n = 126; 77.8%) were enrolled at primary diagnosis, whereas INFORM analysis was performed due to refractory disease or progression under first-line treatment in 15 patients. For the 10 patients registered at first relapse (after completion of first-line treatment) the median latency from primary diagnosis to relapse was 10.5 months (range 0.9–47.9 months), whereas eight patients suffered from second or multiple relapses with a median latency of the current episode from primary diagnosis of 29.9 months (range 7.5–91.4 months). In 60.5% of patients, only a biopsy was performed at the current episode to obtain tumor material, with the remainder undergoing some form of resection. Details on clinical characteristics for the whole cohort as well as separated for primary and relapse/progressive disease are provided in Table 1.
Table 1 Details on clinical features of the study cohortDNA methylation subgrouping of DMGGenome-wide DNA methylation analysis was performed on 160 tumors and it was not possible for two tumors. Based on methylation profiling using the current version v12.8 of the “Heidelberg classifier” (https://app.epignostix.com), 155 tumors (96.9%) were allocated to the group “DMG H3 K27-altered” (DMG_K27) with 134 tumors having a methylation score > 0.9 for the DMG_K27 group. A total of 149 tumors of the DMG_K27 subgroup harbored the characteristic mutation at position 27 of one of the histone 3 isoforms (histone 3.3 in 129 tumors and histone 3.1 in 20 tumors), leading to lysine to methionine substitution (K27M). Of the six H3 K27-wildtype tumors in this subgroup, overexpression of EZHIP was observed in three tumors, one had an EZH2 single-nucleotide variant and two tumors featured none of these alterations.
Five tumors were classified as “DMG with EGFR alteration” (DMG_EGFR), of which four harbored an EGFR alteration (single nucleotide variant (SNV), small insertion/deletion (InDel) or amplification), with three of these displaying additional EZHIP overexpression and one H3.1 K27M-mutation, respectively. No EGFR alteration, but H3.3 K27M mutation was found in the fifth tumor of the DMG_EGFR subgroup.
Visualization by t-stochastic neighbor embedding (t-SNE) was used for comparison of the current cohort with a large CNS tumor reference cohort published by Capper et al. [7] (Fig. 1). All tumors from the current INFORM cohort formed a clear cluster with the DMG K27-altered group of the Capper reference cohort, confirming the molecular subgroup allocation. Within the DMG_K27 cluster on t-SNE, tumors with histone H3.1 K27M-mutation fell together at the edge of the larger cluster. Further, tumors of the DMG_EGFR subgroup with EGFR alteration formed a small subcluster.
Fig. 1
Clustering based on whole-genome DNA methylation analysis visualized by t-stochastic neighbor embedding (t-SNE) combined with reference cohort from Capper et al. [7]. DMG_K27 Capper = methylation class diffuse midline glioma with Histone 3 K27-alteration included in cohort from Capper; DMG_K27/K27-wt = methylation class diffuse midline glioma Histone 3-altered, without K27M-mutation; DMG_K27/H3.3 K27-mut = methylation class diffuse midline glioma with Histone 3.3 K27M-mutation; DMG_K27/H3.1 K27-mut = methylation class diffuse midline glioma with Histone 3.1 K27M-mutation; DMG_K27/EZHIP-EZH2 = methylation class diffuse midline glioma Histone 3-altered, with EZHIP overexpression or EZH2 alteration; DMG_EGFR/EGFR-alt = methylation class diffuse midline glioma EGFR-altered, with EGFR alteration; DMG_EGFR/EGFR-with = methylation class diffuse midline glioma EGFR-altered, without EGFR alteration
Pathways altered in DMGThe p53 pathway, which is characteristically altered in pedHGG and DMG [33], was also frequently affected in our cohort, with 100 tumors (61.7%; 99 tumors with SNV or InDel, one tumor with intragenic rearrangement) harboring TP53 alterations, 7 tumors ATM mutations and 13 tumors featuring alterations of PPM1D (Fig. 2). Alterations in genes coding for receptor tyrosine kinases (RTKs) were identified in PDGFRA (n = 34), followed by FGFR1 (n = 11), MET (n = 11) and EGFR (n = 5). Notably, co-amplification of the nearby located genes KDR and KIT together with PDGFRA amplification—which is a well-known finding in HGG and other tumor entities [8, 16, 35]—was present in eleven tumors. The PI3K-AKT-MTOR pathway was affected most frequently by PIK3CA alterations (n = 31) as well as PIK3R1 (n = 9) and PTEN (n = 10). Besides being the pathognomonically altered pathway in pediatric low-grade glioma, alterations in the MAPK pathway (downstream of RTKs) are also described in pedHGG [33]. In the cohort presented here, we identified BRAF V600E mutations in five tumors as well as alterations in e.g. NF1 (n = 14) and KRAS (n = 5). Moreover, 22 tumors (13.6%) harbored alterations in genes playing a role in cell cycle control, e.g. CDKN2A/B (n = 4), CDKN2C/CDKN2D (n = 6), CDK4 (n = 5) and CCND1/CCND2 (n = 11) (alterations not mutually exclusive). Further DMG-typical findings included alterations in ATRX (n = 30) and ACVR1 (n = 22). Whereas, less common in DMG but occasionally observed in our cohort were, for example, MYC/MYCN (n = 8) and BCOR (n = 9) alterations. Comparing tumors analyzed at primary diagnosis versus relapsed/progressive tumors, most alterations occurred at a similar frequency during the disease course. PDGFRA was more often affected in the tumors at primary diagnosis (24.6% vs 9.1% at relapse/progression), whereas ACVR1 mutations were identified in 27.3% of tumors at relapse/progression compared to 10.3% at primary diagnosis. Four of the five detected BRAF V600E mutations were found in relapsed/progressive tumors (12.1% vs 0.8% at primary diagnosis). With regard to metastatic status, no clear differences in alteration frequencies were observed. Tumors of patients with M0 situation (n = 123) showed a slightly lower frequency of ALT positivity (27.6%) compared to tumors of patients with metastatic disease (any M+, n = 14; 50% ALT positive; p = 0.12), as well as more rarely alterations in the MAPK pathway (13.0% in M0 versus 35.7% in M+; p = 0.04) and in genes of cell cycle control (11.4% in M0 versus 28.6% in M+; p = 0.09). The cohort included tumors taken both before and after initiation of antitumor treatment, including radiotherapy. Even though alteration frequencies did also not obviously vary between radiotherapy-naїve (n = 130) and radiotherapy-exposed (n = 29) tumors, alterations in PDGFRA and ATRX occurred more often in tumors taken before radiotherapy (23.8% PDGFRA-altered and 20.8% ATRX-altered) compared to tumors taken after radiotherapy (10.3% PDGFRA-altered; p = 0.14 and 10.3% ATRX-altered; p = 0.29). On the other hand, there was a significant difference in the frequency of alterations in cell cycle control, which were found in 9.2% of tumors taken before and in 27.6% of tumors taken after radiotherapy (p = 0.01).
Fig. 2
Clinical characteristics and molecular alterations of 162 diffuse midline glioma cases. H3 = Histone 3; ALT = alternative lengthening of telomeres; NA = not available; M0 = no evidence of tumor dissemination; M1 = microscopic presence of tumor cells in CSF; M2 = gross tumor dissemination in subarachnoid space or ventricles; M3 = gross nodula seeding in spinal subarachnoid space; M+ = metastatic disease but extend of metastatic spread unknown; R4 = biopsy; R3 = partial resection; R2 = rim near total resection; R1 = gross total resection; R+ = residual tumor but extent of resection unknown; DMG_K27 = methylation class diffuse midline glioma Histone 3 K27-altered; DMG_EGFR = methylation class diffuse midline glioma EGFR-altered; H3.3 K27 = Histone 3.3 K27M-mutation positive; H3.1 K27 = Histone 3.1 K27M-mutation positive; H3 K27 + G34 = H3.3 K27 = Histone 3.3 K27M-mutation and G34-mutation positive; InDel(del) = small deletion; InDel(ins) = small insertion; ITD = internal tandem duplication
Especially with regard to the unfavorable prognosis of DMG patients, the identification of actionable targets in the respective tumors is essential. The analysis of the current cohort revealed genetic alterations that were considered as a potential target for targeted therapy in 131 tumors (80.9%), with alterations in e.g. PDGFRA, FGFR1, EGFR and MET, KRAS, BRAF, PIK3CA and CDK4 being considered as priority targets. Together, alterations with a priority of ‘very high’ or ‘high’ according to the INFORM scoring system [51] were detected in 40 tumors (24.7%).
The median tumor mutational burden (TMB) was 0.49 somatic mutations per Megabase (Mb) (range 0.09–9.01). Thus, no tumor of the current cohort showed a hypermutator phenotype with a cut-off being set at TMB > 10 somatic mutations per Mb [6].
In 46 tumors (28.4%), a positive genomic signature for alternative lengthening of telomeres (ALT) was observed. Half of these tumors harbored ATRX alterations (23/30 ATRX-altered tumors).
Likely pathogenic germline alterations, which were considered to be potentially relevant for the current tumor disease were identified in six cases, namely affecting the genes CHEK2 (n = 2), PALB2, SLX4, MUTYH, NBN (each n = 1). Whether these are truly playing a tumor driver role in this context, however, remains an open question. According to the information provided in the eCRF none of these alterations were known before in the respective patients’ medical history.
Characteristics of specific subgroupsThe most frequent tumor localization after pons/brainstem in the current cohort was the thalamus (n = 19). Median age at primary diagnosis of patients suffering from a thalamic DMG was slightly older compared to the whole cohort at 11 years (range 3–17 years). Molecularly, thalamic DMG were assigned to the DMG_K27 cohort in 17 tumors, with 14 of these harboring an H3.3 K27M mutation, whereas two tumors belonged to the DMG_EGFR subgroup. Almost half of the thalamic tumors had a positive genetic signature for ALT and notably, all three tumors with an EZHIP overexpression were located in the thalamus. Median overall survival (OS) of the current episode for patients with thalamic DMG was 14.0 months (range 4.5–31.8 months) with five patients being alive at last follow-up (no follow-up data available for two patients).
The nine patients with DMG located in the spinal cord were even older at primary diagnosis (median age 13 years, range 5–16 years). These tumors all belonged to the molecular DMG_K27 subgroup featuring the typical K27M mutation in histone H3.3. All patients of this group died of disease after a median OS of the current episode of 8.1 months (range 2.2–65.1 months).
One further specific molecular subgroup are the 21 tumors harboring a histone H3.1 K27M mutation. The respective patients were slightly younger at primary diagnosis compared to the whole cohort with a median age of 5 years (range 3–15 years). All except one patient were enrolled at primary diagnosis or with refractory/progressive disease. Most of these tumors were located in the pons or brainstem (n = 16; 76.2%). Median OS from primary diagnosis for this subgroup was 16.5 months (range 0.1–26.6 months) and three patients were alive at last follow-up. Notably, 76.2% of the H3.1 mutated tumors featured a concomitant ACVR1 mutation, BCOR alterations were identified in 6 tumors (29%) and all tumors of this subgroup were negative for the ALT signature as well as for ATRX alterations.
Another interesting subgroup are tumors with alterations in the MAPK pathway (n = 27). Median age at primary diagnosis for these patients was 11 years (range 3–17 years) and median OS of the current episode 15.8 months (range 1.7–65.1 months). An H3.3 K27M mutation was detected in 25 tumors and H3.1 K27M mutation in one tumor. Within this group of MAPK-altered DMG, five tumors harbored a BRAF V600E mutation, which is a typical characteristic for lower-grade gliomas. One patient from each of these five patients was enrolled in the INFORM registry at primary diagnosis, first, second and third relapse, respectively, or due to refractory disease. Tumor localization was in the brainstem in one patient and the thalamus in two patients (no clear localization in two patients) and two patients had metastatic disease at the time point of INFORM enrollment. Molecularly, all five tumors were assigned to the DMG_K27 subgroup by methylation analysis and all harbored an H3.3 K27M mutation. One of the tumors featured an ALT-positive status and a potential pathogenic germline alteration was identified in two of these five patients (namely CHEK2 and SLX4). After molecular analysis of the tumors through the INFORM pipeline, three patients received radiotherapy in combination with temozolomide (plus valproic acid in one patient). Two of these patients were treated with trametinib plus dabrafenib in addition. One patient received vemurafenib, cyclophosphamide and chloroquine (no data on treatment available for one patient). Tumor progression occurred in three patients after a median of 24.9 months (range 6.3–25.2 months) after diagnosis of the current episode (no data on progression available for two patients). Four patients died of the disease with a median survival of 14.6 months (1.7–37.4 months) after diagnosis of the current episode and 37.5 months (13.4–72.4 months) after primary diagnosis. One patient was alive at the last follow-up 24.9 months after diagnosis of the current episode and 28.1 months after primary diagnosis.
For the tumors assigned to the DMG_EGFR subgroup (n = 5), as well as the tumors with EZHIP overexpression or EZH2 mutation (n = 4) and the two tumors without histone 3 K27 mutation or EZHIP/EZH2 alteration within the DMG_K27 subgroup, small numbers hindered to make any reasonable comparisons.
Gene expression analysisGene expression profiling based on RNA sequencing was available for 123/162 DMG. Comparison of differentially expressed genes between these samples of the current DMG cohort and 67 samples of non-DMG, histone 3 wild-type pedHGG patients from the INFORM registry showed a clear separation of the two groups based on z-Scores (Fig. 3a, b). The 500 most differentially expressed genes included exemplarily NTRK2 (Supplementary Fig. 2a).
Fig. 3
Differential gene expression comparing DMG and other pedHGG as well as several subgroups within the DMG cohort. a Heatmap with 500 most differentially expressed genes comparing 123 DMG and 67 non-midline, H3-wildtype pedHGG tumors shows separation of the two groups. For volcano plots, genes with high fold changes (log2 fold change; x-axis) and high statistical significance (− log10 of p-value; y-axis) are illustrated. b Volcano plot illustrating the most differentially expressed genes in DMG versus pedHGG. Volcano plots c–g comparing differential gene expression between subgroups, based on c current episode (primary diagnosis vs refractory/relapsed disease); d tumor localization (pons/brainstem vs thalamus); e MAPK pathway alteration status (alteration in MAPK pathway vs no alteration in MAPK pathway); f TP53 alteration status (TP53-altered vs TP53-wildtype); g Histone H3 mutation status (Histone H3.1 K27M mutation vs Histone H3.3 K27M mutation). DMG diffuse midline glioma, pedHGG pediatric-type high-grade glioma, expr expression
Furthermore, different subgroups within the DMG cohort were investigated and compared: current episode (primary diagnosis versus refractory/relapsed disease; Fig. 3c), tumor localization (pons/brainstem versus thalamus; Fig. 3d), MAPK pathway alteration status (MAPK pathway altered versus no MAPK pathway alteration; Fig. 3e), TP53 alteration status (TP53-mutated versus TP53-wildtype; Fig. 3f) and Histone H3 mutation status (Histone H3.1 K27M mutation vs Histone H3.3 K27M mutation; Fig. 3g). Regarding tumor localization differentially expressed genes included OTX2 (Supplementary Fig. 2b). Expression of ALK, SETD2, ACVR1B and RAF1 (Supplementary Fig. 2c–f) as some examples was lower in tumors harboring a Histone H3.1 K27M mutation compared to tumors with Histone H3.3 mutation.
Gene set enrichment analysis applying the ‘oncogenic signature gene sets’ of the Human MSigDB Collections revealed enrichment in DMG compared to pedHGG of genes regulated upon expression changes of PTEN, KRAS, MYC and ATM as some examples (Supplementary Fig. 3a–d, Supplementary Table 1).
Treatment dataData on treatment of the current episode were available for 153 patients (94.4%), previous treatment (if applicable) for 35 patients and outcome data for 145 patients (89.5%; not available n = 17), respectively.
The majority of patients enrolled at primary diagnosis (n = 126, including 3 patients with no treatment data available) were treated with radiotherapy (n = 105; 85.4%), with nine of these patients receiving no additional systemic therapy. Besides the widely applied systemic therapy with temozolomide and/or valproic acid (n = 89), other chemotherapeutic agents were applied in 11 patients, tyrosine kinase inhibitors in 40 patients, other targeted therapeutic agents in six patients, immunotherapy in two patients and other treatments such as chloroquine or methadone in eight patients (several patients received multiple treatments). A total of 55 patients enrolled at primary diagnosis (44.7%) received at least one further therapeutic element in addition or instead of the standard of care treatment (radiotherapy plus temozolomide). No further treatment after surgery was applied to ten patients.
Regarding treatment regimen applied for the current episode in patients with relapsed/refractory disease (n = 33, including four patients with no data on current treatment available), 14 patients were treated with radiotherapy (42.4%); of these nine received re-radiotherapy. Systemic therapy consisted of temozolomide (with or without valproic acid) in nine patients, other chemotherapeutic therapies in nine patients, tyrosine kinase inhibitors in 17 patients, other targeted therapies in four patients, immunotherapy in one and other therapies in three patients. Several patients were treated with different approaches and three patients did not receive any further treatment. Treatment of previous disease episodes in this group of patients included radiotherapy or various systemic therapy approaches, respectively, in 28/33 patients each. Eight patients had received tyrosine kinase inhibitors (n = 7) or immunotherapy (n = 1). Three patients had been treated with radiotherapy only and two other patients enrolled at first relapse were treatment-naїve besides surgery.
Targeted therapy based on molecular profilingA total of 64 patients (41.8% of patients with data on treatment available) were treated with targeted therapies (TT; including immunotherapy and tumor-specific vaccination) after enrollment in the INFORM registry. A list of all applied TTs is provided as Supplementary Table 2. Most of these patients received one TT (n = 32) or two different TTs (n = 21). Three TTs were applied in six patients, four TTs in three patients and treatment included five different TTs in two patients. In total, 114 treatments with TT were initiated in the whole cohort. In 41 patients treated with a total of 51 TTs, the TT was considered matching to respective genetic alterations identified in the INFORM analysis (ten patients were treated with two matching TTs). Thirty of these TTs were directly targeting the respective alteration, whereas the affected pathway was targeted further downstream with 21 TTs. The respective genetic alterations affected the following genes (in some patients several TTs were applied targeting the same alteration): PDGFRA (n = 8), EGFR (n = 2), MET (n = 5), FGFR1 (n = 3), NRAS/KRAS (n = 2), BRAF (n = 4), NF1 (n = 4), MAPK3 (n = 1), PIK3CA (n = 13), PTEN (n = 1), CDK4 (n = 1), CCND1 (n = 1), NTRK (n = 1), TSC1 (n = 1) and H3 vaccine (n = 1). Notably, alterations of KDR/KIT (concomitant with PDGFRA) were additionally targeted in four patients with tumors harboring PDGFRA/KDR/KIT co-amplification.
The most frequently applied TT was ONC201, a dopamine receptor 2 antagonist, which was applied in 19 patients. However, this was not considered to be a particular match to a corresponding alteration due to uncertainties about biomarkers/mechanism-of-action for this compound. The mTOR inhibitor everolimus was part of the therapeutic concept in 13 patients, with genetic alterations in the mTOR pathway being identified in the tumors of eight of these patients. At a similar frequency, trametinib (MEK inhibitor), was used in 12 patients—in two-third of cases (n = 8) in line with alterations affecting the MAPK pathway. Sirolimus (mTOR inhibitor) and paxalisib (PI3K inhibitor) were each prescribed in seven patients. Eight and seven patients, respectively, received bevacizumab (VEGF inhibitor) and nivolumab (checkpoint inhibitor) without a corresponding alteration identified in the respective tumor.
OutcomeFollow-up data on the further clinical course after the current episode with enrollment in the INFORM registry were available for 145 patients (no follow-up data available for 17 patients, two patients excluded from respective analysis due to missing date of primary diagnosis, one patient due to missing date of diagnosis of current episode). Three patients enrolled at primary diagnosis were lost to follow-up less than 6 months after INFORM analysis (still included in survival analysis).
Disease progression after enrollment in the INFORM registry was documented in 103 patients, with a median time to progression of 7.4 months (range 1.8–28.7) from diagnosis of the current event (data on progression missing for 16 patients, five patients died before the first response assessment scheduled 3 months after INFORM analysis). In total, 124/145 patients deceased from the disease during the follow-up period. Median overall-survival (OS) time of these patients from primary diagnosis was 12.9 months (range 1.2–98.5) and 11.0 months (range 1.2–65.1) from diagnosis of the event leading to the enrollment in the INFORM registry. Twenty-one patients were alive at last follow-up, with a median time from diagnosis of the current event of 15.5 months (range 0.1–29.2).
Outcome analysis using Kaplan–Meier estimator revealed a 2-year overall survival (2y-OS) rate for the whole cohort of 10.5% from diagnosis of the current episode and 16.9% from primary diagnosis.
Seventeen patients survived more than 24 months from primary diagnosis (median OS from primary diagnosis 32.7 months, range 26.5–98.5). Seven of these patients were enrolled in INFORM at primary diagnosis, four patients each at first and second or further relapse, whereas refractory/progressive disease under first-line treatment lead to enrollment in INFORM in two patients. One of these patients was primarily diagnosed with an oligoastrocytoma °III more than 7 years preceding the current diagnosis of high-grade glioma. For another patient diagnosis of tuberous sclerosis with giant cell astrocytoma was documented in the medical history—however, no respective germline alteration was identified in the INFORM analysis. A potentially pathogenic germline alteration was identified in three patients of this group. Molecularly, 16 tumors were assigned to the DMG_K27 subgroup with 15 tumors harboring a histone 3 K27M mutation. Notably, the tumors of four longer-term survivors harbored a BRAF V600E mutation.
For survival analysis from primary diagnosis there was a slightly better outcome observed for patients with tumor localized in the spinal cord by univariate analysis (HR 0.396, p-value 0.026, 95% CI 0.175–0.897) and thalamus/basal ganglia (HR 0.555, p-value 0.046, 95% CI 0.311–0.991) compared to pons/brainstem (Fig. 4a). Notably, alterations in the MAPK pathway as well as TP53 alterations were associated with a significant difference in survival (Fig. 4c + d, Supplementary Table 3) whereas patients’ outcome did not differ significantly according to presence or type of histone H3 K27M mutation (Fig. 4b). However, in multivariate analysis only TP53 status was confirmed as a risk factor (TP53 alteration HR 1.888, p-value 0.0015, 95% CI 1.275–2.796; MAPK alteration HR 0.637, p-value 0.107. 95% CI 0.368–1.103; spinal localization HR 0.419, p-value 0.035, 95% CI 0.186–0.943). This is likely due to an interrelationship between the two variables—with MAPK-altered tumors being more likely to be TP53-wildtype. Investigating outcome according to MAPK pathway alteration status only in patients with TP53-wildtype tumors (n = 53) confirmed the difference in survival in association with the presence of MAPK pathway alteration (Supplementary Fig. 4a; HR 0.395, p-value 0.03, 95% CI 0.170–0.922).
Fig. 4
Outcome analysis of 145 DMG patients. a Overall survival (OS) according to tumor localization (from primary diagnosis); b OS according to Histone 3 K27M mutation status (from primary diagnosis); c OS according to MAPK pathway alteration (from primary diagnosis); d OS according to TP53 alteration status (from primary diagnosis); e OS according to disease episode (from diagnosis of current episode); f OS according to general treatment with targeted therapy (from diagnosis of current episode); g OS according to treatment with targeted therapy based on respective target identified (from diagnosis of current episode); h OS according to treatment with tyrosine kinase inhibitor based on respective target (from diagnosis of current episode). NA not applicable, H3.1 histone 3.1, H3.3 histone 3.3, wt wildtype, TT targeted therapy, TKI tyrosine kinase inhibitor
For the evaluation of patients’ outcome according to the treatment applied survival analysis was performed from the diagnosis of the current event. No significant difference was observed according to ‘type’ of current episode (primary diagnosis vs relapsed/refractory/progressive disease, Fig. 4e, Supplementary Table 3) as well as stratified by treatment with targeted therapy in general or based on priority of respective targets identified in the INFORM analysis (Fig. 4f + g, Supplementary Table 3). However, when comparing treatment with no or not matching TTs to treatment with matching TTs a significant difference in patients’ outcome was observed (Supplementary Fig. 4b, Supplementary Table 3). The same holds true when comparing survival from primary diagnosis in patients receiving standard-of-care treatment only, namely radiotherapy plus temozolomid ± valproic acid, and patients receiving matching targeted therapy anytime during the clinical course (Supplementary Fig. 4, Supplementary Table 3). For the treatment with tyrosine kinase inhibitors (Fig. 4h, regardless of other TTs) as well as for the treatment with ONC201 (Supplementary Fig. 4d, Supplementary Table 3) there seems to be a trend towards better outcome. To investigate whether the observed difference in outcome according to MAPK pathway alteration status was related to the treatment with respective TT, survival analysis was performed for the group of patients with MAPK-altered tumors only (n = 21). Also in this group, no significant difference in outcome was observed according to treatment with TT (matching/not matching to the MAPK alteration) compared to no treatment with TT (Supplementary Fig. 4e; Supplementary Table 3).
Comments (0)