Cell culture and reagents
All cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. HEK293T (RRID:CVCL_0063), HeLa (ATCC Cat# CCL-2, RRID:CVCL_0030), A549 (RRID:CVCL_0023), and HEK293 (RRID:CVCL_0045) cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA). HCT116 (RRID:CVCL_0291) and U2OS (RRID:CVCL_0042) cells were grown in McCoy's 5A medium (Thermo Fisher Scientific, Waltham, MA, USA). U87MG (RRID:CVCL_0022) cells were maintained in Eagle’s minimum essential medium (Nacalai Tesque, Kyoto, Japan). hTERT-RPE1 (RRID:CVCL_4388) cells were maintained in Dulbecco’s modified Eagle medium/nutrient mixture F-12 (Thermo Fisher Scientific, Waltham, MA, USA). All media were supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Nacalai Tesque).
The reagents used in this study included MG132 (474790, Merck, Darmstadt, Germany), carfilzomib (17554, Cayman Chemical, Ann Arbor, MI, USA), bafilomycin A1 (11038, Cayman Chemical), chloroquine (C6628, Sigma-Aldrich), bortezomib (SIH-328, StressMarq Biosciences, British Columbia, Canada), and GSK2830371 (S7573, Selleck Biotechnology, Kanagawa, Japan).
Cloning of cDNAs and plasmid construction
Complementary DNAs encoding the human PPM1D gene were cloned into pGEX-6P-1 (GE Healthcare, Uppsala, Sweden). Wild-type PPM1D, PPM1D-N, PPM1D-C, PPM1D (450–605), PPM1D (502–605), PPM1D (396–570), and PPM1D (396–530), each with an N-terminal FLAG tag, were subcloned into the pQCXIP vector (Takara, Shiga, Japan). Wild-type NQO1 was subcloned into a modified pQCXIP vector (Takara) in which the puromycin resistance gene was replaced with a blasticidin resistance gene.
Retrovirus expression system
Retroviral vectors were transfected into HEK293T cells together with the pCL10A1 vector (Novus Biologicals, Littleton, CO, USA) to generate recombinant retroviruses. HEK293T cells were infected with the recombinant retroviruses in the presence of polybrene (8 μg/ml, Sigma-Aldrich) and selected in a medium containing puromycin (5 μg/ml, Thermo Fisher Scientific) or blasticidin S (10 μg/ml, Fujifilm Wako, Osaka, Japan).
Transfection and immunoblot analysis
Immunoblot analysis was performed with primary antibodies, horseradish peroxidase-conjugated secondary antibodies to anti-Mouse IgG antibody (Promega Cat# W4021, RRID:AB_430834, Madison, WI, USA, 1:15,000 dilution) or anti-Rabbit IgG antibody (Promega Cat# W4011, RRID:AB_430833, 1:15,000 dilution), and an enhanced chemiluminescence reagents (Thermo Fisher Scientific). The following primary antibodies were used: anti-FLAG M2 antibody (Sigma-Aldrich Cat# A8592, RRID:AB_439702, 1:2000 dilution), anti-PPM1D (Cell Signaling Technology, Danvers, MA, USA, 1:2000 dilution), anti-PSMD14 (Proteintech Cat# 12059-1-AP, RRID:AB_2170454, Rosemont, IL, USA, 1:2000 dilution), anti-PSME3 (Proteintech Cat# 14907-1-AP, RRID:AB_2171098, 1:2000 dilution), anti-c-Myc (Cell Signaling Technology Cat# 5605, RRID:AB_1903938, 1:2000 dilution), anti-NCOA4 (Bethyl Cat# A302-272A, RRID:AB_1850160, Montgomery, TX, USA, 1:2000 dilution), anti-vinculin (Sigma-Aldrich Cat# V9131, RRID:AB_477629, 1:50,000 dilution), anti-GAPDH (Thermo Fisher Scientific Cat# AM4300, RRID:AB_2536381, 1:100,000 dilution), anti-NCOA3/SRC3 (Cell Signaling Technology Cat# 2126, RRID:AB_823642, 1:2000 dilution), anti-CDKN1A/p21 (Cell Signaling Technology Cat# 2947, RRID:AB_823586, 1:2000 dilution), anti-TP53 (Cell Signaling Technology Cat# 2524, RRID:AB_331743, 1:2000 dilution), and anti-NQO1 (Proteintech Cat# 11451-1-AP, RRID:AB_2298729, 1:2000 dilution).
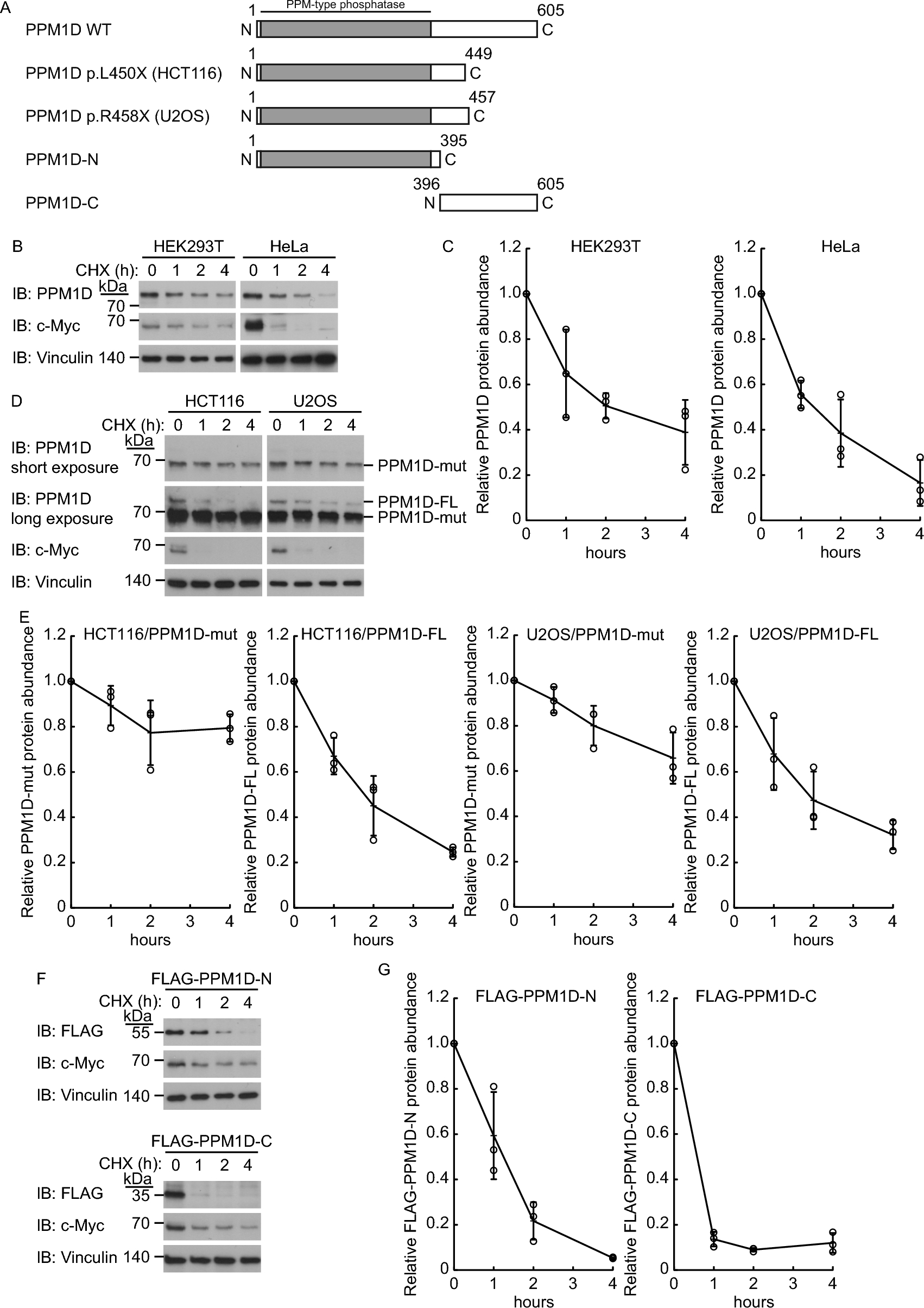
Cycloheximide chase assay
Cycloheximide (50 μg/ml, Sigma-Aldrich) was added to the culture medium to inhibit translation and cell lysates were prepared at the indicated time points. The cell lysates were examined using immunoblot analysis. Densitometric analysis was carried out using NIH-ImageJ software.
Sample preparation for mass spectrometry analysis
HEK293T cells (4.0 × 108) stably expressing FLAG-tagged PPM1D-C were lysed in a solution containing 50 mM Tris–HCl (pH 7.6), 300 mM NaCl, 10% glycerol, 0.2% NP-40, 10 mM iodoacetamide (Sigma-Aldrich), 10 mM N-ethylmaleimide (Sigma-Aldrich), 0.5 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF, Roche, Branchburg, NJ, USA), and 10 μM MG132 (Merck, Darmstadt, Germany). The cell lysates were sonicated and centrifuged at 20,000 × g for 10 min at 4 °C, and the resulting supernatant was incubated with anti-FLAG M2 agarose (Sigma-Aldrich) for 2 h at 4 °C. The resin was separated by centrifugation, washed five times with ice-cold lysis buffer and eluted twice with 50 μl of lysis buffer containing 0.25 mg/ml FLAG peptide (Sigma-Aldrich). Eluted peptides were dried by vacuum centrifugation, reduced with 55 mM dithiothreitol at 95 °C for 5 min (Thermo Fisher Scientific), alkylated with 10 mM iodoacetamide for 20 min at room temperature in the dark (Thermo Fisher Scientific), and digested overnight at 37 °C with 50 ng/μl trypsin (Promega, Madison, WI, USA) in the presence of 0.01% RapiGest SF (Waters, Milford, MA, USA). After tryptic digestion, the samples were acidified with TFA and purified by solid-phase extraction using GL-Tip GC and GL-Tip SDB (GL Sciences, Tokyo, Japan).
Mass spectrometry analysis
Desalted tryptic digests were analyzed by nano-flow ultra-high-performance liquid chromatography (EASY-nLC 1000; Thermo Fisher Scientific) coupled online to an Orbitrap Elite instrument (Thermo Fisher Scientific). The mobile phases were 0.1% formic acid in water (solvent A) and 0.1% formic acid in 100% acetonitrile (solvent B). Peptides were directly loaded onto a Reprosil-Pur C18 analytical column (3 μm in particle size, 75 μm in inner diameter, and 12 cm in length; Nikkyo Technos, Tokyo, Japan) and separated using a 150-min two-step gradient (0–35% for 130 min, 35–100% for 5 min, and 100% for 15 min of solvent B) at a constant flow rate of 300 nl/min. For ionization, a liquid junction voltage of 1.6 kV and a capillary temperature of 200 °C were used. The Orbitrap Elite instrument was operated in the data-dependent MS/MS mode using Xcalibur software (RRID:SCR_014593, Thermo Fisher Scientific) with survey scans acquired at a resolution of 120,000 at m/z 400. The top 10 most abundant isotope patterns with a charge ranging from 2 to 4 were selected from the survey scans with an isolation window of 2.0 m/z and fragmented by collision-induced dissociation with normalized collision energies of 35. The maximum ion injection times for the survey and MS/MS scans were 60 ms, and the ion target values were set to 1 × 10⁶ for the survey and MS/MS scans.
Protein identification from MS data
Proteome Discoverer software (version 2.4; RRID:SCR_014477; Thermo Fisher Scientific) was used to generate peak lists. The MS/MS spectra were searched against the UniProt Knowledgebase (version 2017_10; RRID:SCR_002380) using the SequestHT search engine. The precursor and fragment mass tolerances were set to 10 ppm and 0.6 Da, respectively. Methionine oxidation, protein amino-terminal acetylation, Asn/Gln deamidation, Ser/Thr/Tyr phosphorylation, diglycine modification of Lys side chains, and Cys carbamidomethyl modification were set as variable modifications for database searching. Peptide identification was filtered at a 1% false discovery rate. Label-free quantification was calculated using the intensities of precursor ions in the precursor ions quantifier node. Normalization was performed such that the sum of the abundance values for all peptides in each sample was the same. The log2 value of the abundance ratio (PPM1D-C versus Mock) was calculated for proteins related to proteasome subunit.
RNA interference
Silencer Select siRNA for human PSMD14 (s19921 and s19919), human PSME3 (s19871 and s19872), and negative control (4390843) were purchased from Thermo Fisher Scientific. HEK293T cells were transfected with the siRNA using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific).
Recombinant protein purification
Recombinant human PPM1D protein was expressed in Escherichia coli strain DH5α harboring pGEX-6P-1-PPM1D for 16 h with 0.2 mM isopropyl-β-d-thiogalactopyranoside (Sigma-Aldrich). One liter of bacterial culture medium was centrifuged, and the pellet was resuspended in 40 ml of PBS (Thermo Fisher Scientific) supplemented with AEBSF (Roche, Branchburg, NJ) and disrupted with a French pressure cell (Aminco, Thermo Fisher Scientific). The lysates were clarified by ultracentrifugation, and the supernatants were incubated on ice for 2 h after the addition of 1 ml of glutathione sepharose 4B beads (GE Healthcare, Uppsala, Sweden). The beads were washed 5 times in PBS and the bound proteins were treated with PreScission protease (Cytiva, Marlborough, MA, USA) at 4 °C overnight. The excised GST and PreScission protease were removed from PPM1D by repurification on glutathione-Sepharose 4B beads.
Degradation of PPM1D protein by 20S proteasomes
Human 20S proteasome protein was purchased from Boston Biochem (Cambridge, MA, USA). The 20S proteasome (0.25 μg) was incubated with 2 ng PPM1D protein for 1 h at 30 °C in a buffer containing 50 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 1 mM DTT, and 0.2 mg/ml BSA. Products were analyzed by SDS-PAGE and immunoblotting.
Cell viability assay
Cell viability was evaluated using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). 1 × 104 cells in 100 µl of medium were seeded into each well of a 96-well plate and incubated at 37 °C for 48 h with the indicated concentrations of bortezomib, GSK2830371, or both. After incubation, 10 µl of Cell Counting Kit-8 reagent was added to each well, and the cells were incubated for an additional 1 h at 37 °C. The absorbance was then measured at 450 nm using a SpectraMax Paradigm (Molecular Devices, San Jose, CA, USA). The cell viability rate was calculated as a percentage of the control based on samples without drug exposure.
Evaluation of drug combination effects
To evaluate the combination effects of bortezomib and GSK2830371, cell viability data were analyzed using CompuSyn software (ComboSyn Inc., Paramus, NJ, USA), based on the Chou–Talalay method [22]. Drug concentrations were applied at a fixed molar ratio derived from the IC50 values of each single agent. The combination index (CI) was calculated at different effect levels (fraction affected, Fa), where CI < 1 indicates synergism, CI = 1 indicates an additive effect, and CI > 1 indicates antagonism. Median-effect plots CI–Fa plots, and isobolograms were generated automatically by the software.
Establishment of bortezomib-resistant cells
To establish bortezomib-resistant cell Lines, parental cells were continuously cultured for 30 days in medium containing bortezomib at concentrations ranging from 10 to 100 nM, increasing in 5 nM increments. Cells were passaged every 2 to 3 days to maintain sub-confluent growth. After 30 days, cells that survived in the highest bortezomib concentration were collected as the resistant population. Bortezomib resistance was evaluated using a cell viability assay, in the same manner as for the parental cells, by determining the IC50 values after 48 h of bortezomib treatment. Resistance was confirmed by comparing the IC50 values between the resistant and parental cell lines.
Statistical analysis
All experiments were performed at least three independent times or at least twice in triplicate. Data are presented as mean ± standard deviation (SD). The unpaired Student's t-test was used to compare differences between experimental and control groups. Differences with P < 0.05 were considered statistically significant. All analyzes were performed using Microsoft Excel (RRID:SCR_016137).


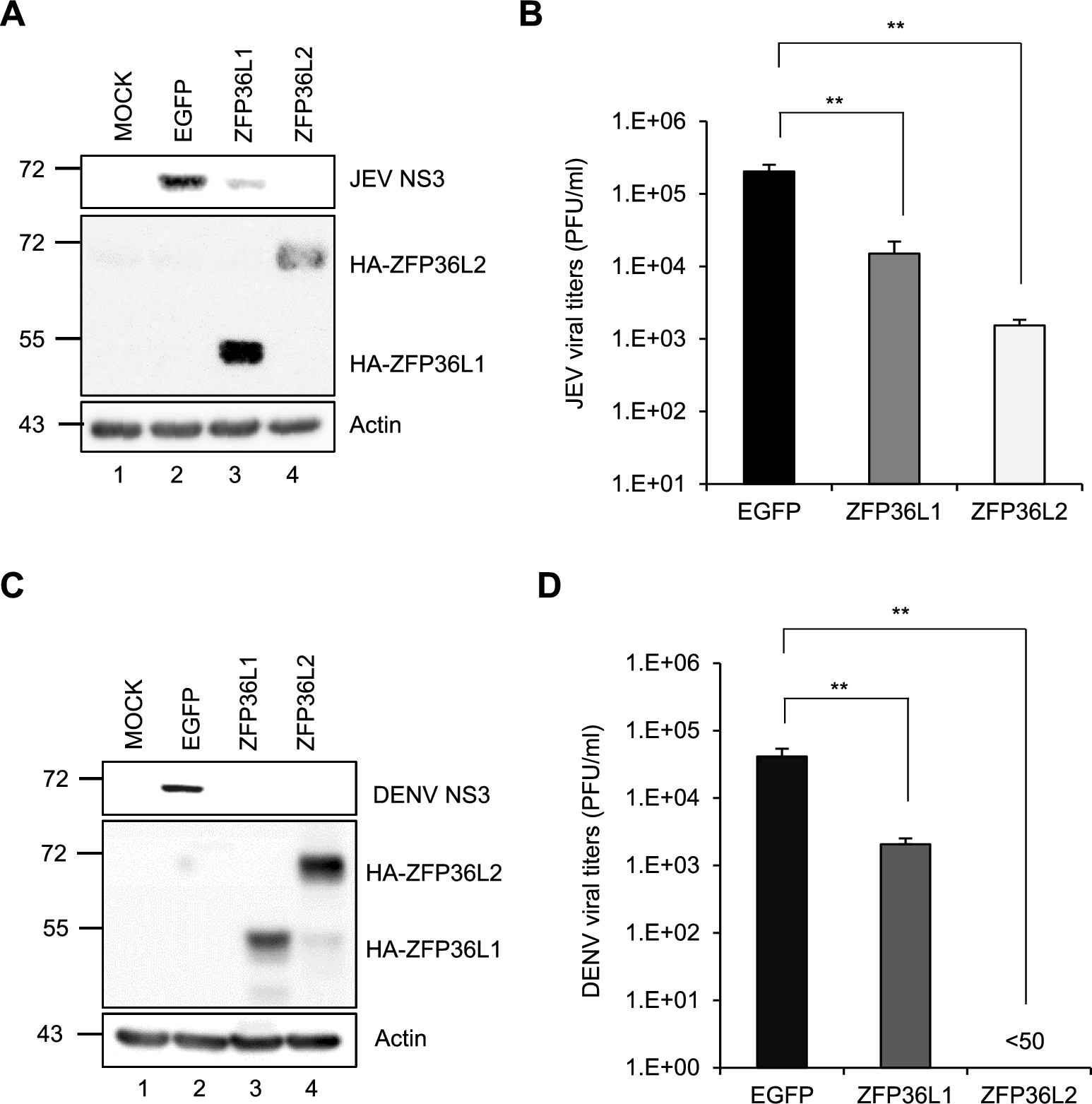
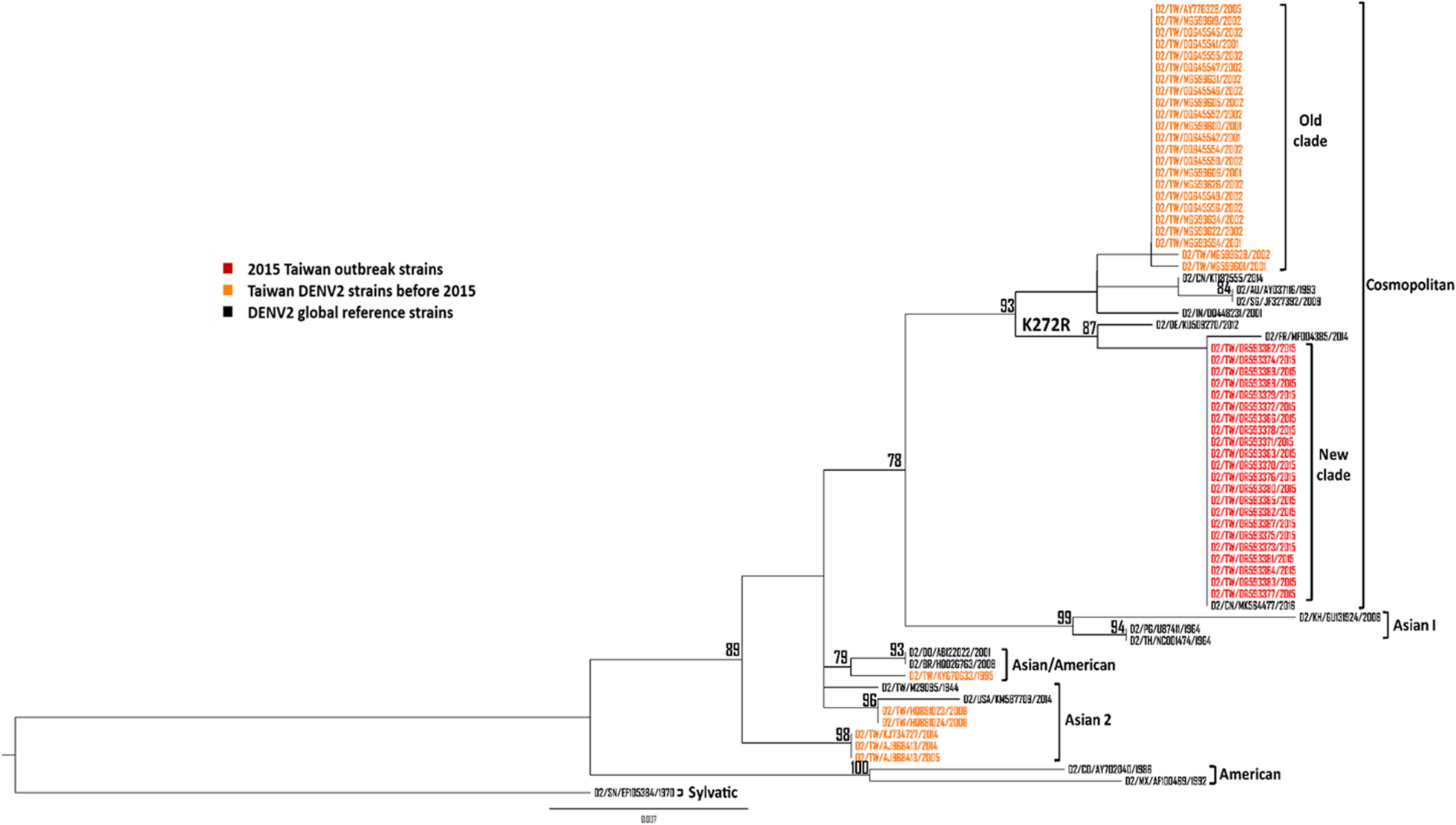



Comments (0)