Cell culture
All cell lines were obtained from ATCC and maintained at 37 ℃ with 5% CO2. Baby hamster kidney (BHK-21) cells (ATCC: CRL-12071) and A549 cells (ATCC: CCL-185) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Hyclone) and 2% penicillin/streptomycin (P/S; Gibco). Vero cells (ATCC: CCL-81) were cultured in Eagle’s Minimum Essential Medium (EMEM; Gibco) supplemented with 10% FBS, 2% P/S, and 1% sodium pyruvate (Gibco), and 293 T cells (ATCC: CRL-3216) were cultured in DMEM supplemented with 10% FBS and 1% sodium pyruvate. For M1 macrophage differentiation, THP-1 cells (ATCC: TIB-202) were first cultured in RPMI medium supplemented with 10% FBS. To induce macrophage differentiation, THP-1 cells (2 × 105/mL) were seeded in 48-well plates and treated with PMA (100 nM) for 20–24 h, followed by a 48h resting period. The cells were then incubated in RPMI medium containing 10% FBS, IFN-γ (20 ng/mL), and LPS (100 ng/mL) for an additional 24 h to achieve M1 polarization.
Phylogenetic analysis
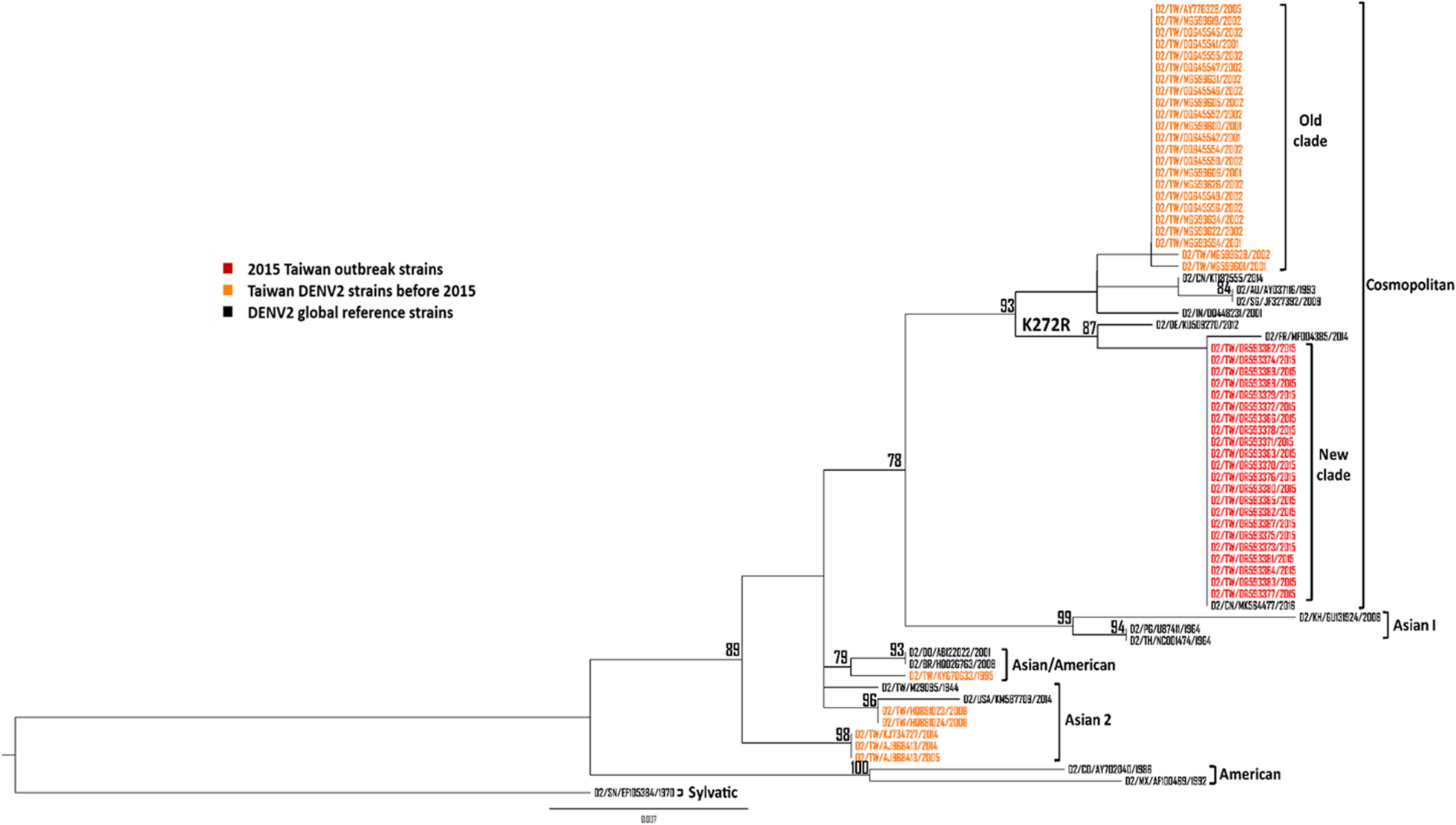
All DENV2 NS1 sequences of Asian countries including China, India, Indonesia, Singapore, Malaysia, Thailand, Vietnam, and the Philippines from 1995 to 2019 available on GenBank were downloaded (accessed on March 1, 2024). Two different sequence datasets were generated for phylogenetic analysis. The first dataset was composed of all Taiwan DENV2 NS1 sequences in GenBank from 1995 to 2014 including 22 NS1 sequences of 2015 Taiwan outbreak strains as well as 17 global reference strains (accession nos. KU509270, MF004385, AY037116, KT87555, JF327392, DQ448231, GU131924, U87411, NC001474, HQ026763, AB122022, KM587709, M29095, AY702040, AF100469, MK564477, and EF105384). We randomly selected representative strains from DENV2 isolates of Asian countries into another dataset, 22 of the 2015 Taiwan outbreak strains, and 17 global reference strains were also included. Phylogenetic tree was inferred from the alignment using the maximum likelihood approach generated in IQ-TREE software version 2.3.3 with bootstrapping by 1000 replicates and visualized using FigTree 1.3.1 programs.
Genetic variation analysis
We previously sequenced 22 DENV2 isolates (accession nos, OR593362-OR593383) from the 2015 outbreak in Taiwan using next-generation sequencing (NGS) [30]. The consensus sequences of each isolate were compared to 29 Taiwan DENV2 strains before 2015 in GenBank to determine the amino acid substitutions of this outbreak.
Construction of infectious cDNA clones
The DENV2 (16681 strain, Accession no.: KU725663.1) was used as template for infectious clone construction. The construction of infectious clones was similar as previously described [30]. In brief, the plasmid pDL-DENV2-EGFP-C60-10062016-A2 was used as template to perform site-directed mutagenesis producing reverse genetics (rg) viruses containing each amino acid substitution isolated from the outbreak. The specific primers used in site-directed mutagenesis were listed in Table S1 The plasmids were transfected into BHK-21 cells (8 × 105/well) in 6-well plates using PolyJet™ in vitro DNA Transfection Reagent (SignaGen) according to manufacturer’s instructions. The rg viruses were harvested at 7–9 days post-transfection when the fluorescence was observed on more than 75% of cells. Then, the rg viruses were passaged twice in Vero cells and collected for further experiments.
Construction of recombinant NS1 clones
The full-length DENV2 NS1 sequence was cloned into pGEM-T vector by TA cloning using the plasmids of WT virus and K272R mutant as template for the amplification of NS1 sequence. The sequences of recombinant NS1 clones were then constructed into pTT5-glu with Kozak consensus sequence in front of the beginning of NS1 sequence which is important for efficient translation initiation. The primers used in the construction of recombinant NS1 clones were listed in Table S1.
Infection and transfection
DMEM (2% FBS, 2% P/S) and EMEM (2% FBS, 2% P/S, 1% sodium pyruvate) viral medium were used in infection experiments to analyze the growth kinetics, viral fitness and stability, ISGs mRNA expression as well as cytokine secretion. A549 cells (6 × 105/well) were seeded in 6-well plates and incubated for 24 h and differentiated M1 macrophage (~ 1 × 105/well) were well differentiated in 48-well plates first. The cells were then infected by rg viruses with a 1 h adsorption period. For the transfection of pTT5-NS1 plasmids, 293 T cells (8 × 105/well) were seeded in 6-well plates for 24 h. The plasmids were transfected into cells using HyFect® Transfection Reagent (Leadgene Biomedical, Taiwan) following manufacturer’s instruction and the old medium was replaced by the FreeStyle™ 293 Expression Medium (ThermoScientific) after 24 h incubation.
IFN-α treatment
The infected or transfected cells were treated with human IFN-Alpha 2b (2000 IU/mL) for 6 h at 3 days post-infection before the sample was collected for quantitative PCR of ISGs including IFIT1, ISG15, and MxA. For the inhibition assay of STAT1 phosphorylation, the infected or transfected cells were treated with human IFN-Alpha 2b (2000 IU/mL) for 1 h before the sample was collected for Western blot analysis.
ELISpot assay for rgDENV quantification
Vero cells (2 × 104/well) were seeded in 96-well plates for 24 h and infected with 30 µL of ½ log serially diluted rg viruses. After 1 h incubation, 150 µL of overlay medium, which contains EMEM viral medium and 1% methyl cellulose, was added into each well. The supernatant was aspirated at 4 days post-infection and the cells were washed with PBS twice. The cells were fixed with 80% methanol and freeze-thawed before immunostaining. The methanol was removed, and the cells were blocked with 5% skimmed milk in PBS. After 30 min of blocking at room temperature, the cells were stained with primary antibodies, mouse anti-dengue complex monoclonal antibody (MAB8705, Millipore) at 1:2000 dilution and incubated at 37 °C for 2 h. Then, the cells were washed 4 times with PBS-T (0.05% Tween 20 in PBS) before staining with goat anti-mouse IgG conjugate HRP (KPL (#5220-0460), LGC Seracare). After 2 h incubation, the cells were washed 6 times with PBS-T and the TrueBlue Peroxidase Substrate (KPL (#5510-0030); LGC Seracare, Milford, MA, United States of America) was added to each well and incubated for 30 min in the dark. The photographs of each well were then taken using the ImmunoSpot system and the focus forming units per milliliter (FFU/mL) was then calculated.
Growth kinetics
A549 cells and Vero cells (2 × 105/well) were seeded in 24-well plates for 24 h and infected by rg viruses at MOI 0.01. The viruses were collected at 0-, 1-, 2-, 3-, and 4-days post-infection then titrated by ELISpot assay.
Viral competition assay
Vero cells (1 × 105/well) were seeded in 24-well plates for 24 h and infected by WT virus, mutant viruses, or 1:1 mixture of WT and each mutant. After each passage, 100 µL each group of viruses were used for the following passage up to P5. Sanger sequencing was then performed to determine the major variant at P1 and P5.
NS1 ELISA
The procedure of this experiment was similar to the commercialized ELISA kits except that the capture antibodies and detection antibodies were in-house products while the standard was purchased additionally. The capture antibody was a 1:1 mixture of 31B2 and 33D2 anti-NS1 monoclonal antibodies, and the detection antibody was biotin-labelled 33D2 anti-NS1 antibody. The working concentration of each antibody were 5 µg/mL and 2 µg/mL, respectively. The standard used in this experiment was purchased from the Native Antigen Company, which is DENV2 NS1 hexamer produced in mammalian HEK293 cells with purity higher than 95% determined by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE).
Quantitative real-time PCR analysis
The infected or transfected cells were treated with human IFN-alpha 2b (Alpha 2) Protein (PBL Assay Science) before the samples were collected for further analysis. The mRNA of infected A549 cells and M1 macrophages were extracted using the Genomic Total RNA Extraction Kit (RBC Bioscience) and then 1 µg of total RNA was reverse transcribed into cDNA using MMLV Reverse Transcriptase (Promega) with random primers. Quantitative real-time PCR (qPCR) was performed with 2 × qPCRBIO SyGreen Blue Mix in StepOnePlus Real-Time PCR System. The primers of IFN-β, ISGs, and pro-inflammatory cytokines used in the qPCR were listed in Table S2.
Enzyme linked immunosorbent assay (ELISA)
The culture supernatants from infected A549 cells and stimulated M1 macrophages were collected to measure cytokine levels. For A549 cells, IFN-I and pro-inflammatory cytokines, including IFN-β, IL-6, and IL-8, were quantified. For M1 macrophages, IL-6 and IL-8 levels were measured. All cytokine measurements were performed using Human DuoSet ELISAs (Bio-Techne) according to the manufacturer’s instructions.
Western blot analysis
The cell lysates were collected, and the total protein levels were quantitated by Bradford assay (BioRad). The pSTAT1 (9172, Cell Signaling Technology), STAT1 (9167, Cell Signaling Technology), pp65 (6956, Cell Signaling Technology), p65 (3303, Cell Signaling Technology), NS1 (LDG0001YA, Leadgene Biomedical), and internal control GAPDH (GTX627408, Genetex) were captured respectively by primary antibodies followed by HRP-conjugated anti-mouse (474-1082, Millipore) or anti-rabbit (7074, Cell Signaling Technology) antibodies. The bound secondary antibodies were then detected by WesternBright ECL HRP substrate (Advansta). The results were quantified using the ImageJ software.
Statistical analysis
All statistical analyses were performed using one-way analysis of variance (ANOVA) or two-way ANOVA in GraphPad Prism 7 software. A value of p < 0.05 represents statistical significance.





Comments (0)