Animals and Chemicals
Eight-week-old MRL/lpr mice were procured from Shanghai SLAC Laboratory Animal Co., Ltd. Female MRL/lpr mice were selected for this study due to the higher prevalence of SLE in females. This murine model demonstrates clinical characteristics that more closely recapitulate human disease manifestations and exhibits more stable disease progression [27]. All animal experiments were conducted in compliance with the guidelines set forth by the Institutional Animal Care and Use Committee of China. After a one-week acclimatization period in our animal facility, the mice were randomly assigned to different experimental groups. The animals were maintained under controlled conditions, with a constant temperature of 25 ± 1 oC, a 12-hour light/dark cycle, and ad libitum access to food. The mice were provided with a standard laboratory diet throughout the duration of the study.
Sodium nitrite (purity ≥ 99.0%, Sigma-Aldrich, 7632-00-0) was administered via drinking water from 9 to 20 weeks of age. Body weights of the mice were recorded weekly. Additionally, lifitegrast (MedChemExpress, HY-19344 A) was administered via intraperitoneal injection once weekly, from 16 to 20 weeks of age.
Experimental Scheme and Sample Collection
The animal experiment in this study consisted of two parts. The first part aimed to investigate the effects of nitrite on MRL/lpr mice. Based on the findings from the first part, the second part of the experiment focused on Itgam to further explore the impact mechanisms of nitrite on lupus.
Experiment I
This experiment aimed to assess the impact of sodium nitrite on lupus development in MRL/lpr mice. The mice were randomly divided into three groups (n = 7 per group): [1] model (MT) group: received free access to drinking water; [2] low-dose nitrite (Low) group: received free access to a 0.1 g/L sodium nitrite solution, with the solution replaced every two days; [3] high-dose of sodium nitrite (High) group: received free access to a 0.3 g/L sodium nitrite solution, with the solution replaced every two days. The experiment was conducted over a 12-week period. Urine samples were collected every two weeks from 12 to 20 weeks of age. Blood samples were collected from the retro-orbital vein and centrifuged at 3000 rpm for 15 min to obtain serum. At 20 weeks of age, the mice were euthanized, and spleen and kidney tissues were harvested. The spleens were immediately weighed and processed into cell suspensions for flow cytometry analysis. One portion of the kidney tissue was snap-frozen in liquid nitrogen and stored at −80 oC, while the other portion was fixed in formaldehyde.
Experimental II
The second part of the study aimed to explore the impact pathogenesis of nitrite on lupus, with a particular focus on integrin alpha M. The MRL/lpr mice were divided into three groups (n = 7 per group): [1] model group: received free access to drinking water; [2] nitrite group: received free access to a 0.3 g/L sodium nitrite solution, with the solution replaced every two days; [3] lifitegrast group: received free access to a 0.3 g/L sodium nitrite solution, and from 16 to 20 weeks of age, mice were administered an intraperitoneal injection of lifitegrast at a dose of 15 mg/kg once weekly. The experiment lasted 12 weeks. Urine samples were collected every two weeks from 14 to 20 weeks of age. Blood samples were obtained from the retro-orbital vein and centrifuged at 3000 rpm for 15 min to obtain serum. At 20 weeks of age, the mice were euthanized to harvest tissues. The spleen was weighed and processed into cell suspensions for flow cytometry analysis. One kidney tissue sample was snap-frozen in liquid nitrogen and stored at −80 oC, while the other was fixed in formaldehyde for further analysis.
Urinary Protein Measurements
To monitor dynamic changes in urinary proteins, urine samples were collected biweekly from each experimental mouse starting at 12 weeks of age. Sample collection was performed using a standard metabolic cage system to ensure uncontaminated, spontaneous urine acquisition. Urinary protein concentrations were quantified using the Bradford Protein Assay Kit (Beyotime, P0006), following the manufacturer’s recommended protocols to maintain measurement consistency and reliability.
Flow Cytometry Analysis
Mouse spleens were homogenized and passed through a 70-mesh filter. Erythrocytes were lysed using an erythrocyte lysis solution, and the lysis was subsequently terminated. Single-cell suspensions were then prepared by adding RPMI 1640 culture medium, and the cell concentration was adjusted to 1 × 10^6 cells/mL. The experiment included blank control tubes, single-staining tubes, and sample tubes. Cells were washed with 1 mL staining buffer, centrifuged, and the supernatant was discarded. A total of 0.5 µL of Fc block antibody was added to all tubes and incubated for 15 min to prevent nonspecific binding. Following incubation, cells were washed again with 1 mL staining buffer, centrifuged, and the supernatant was discarded. For staining, antibodies were added as follows: CD3 (Biolgend, 100204), CD4 (Biolgend, 100412), CD8 (Biolgend, 162308), Bcl-6 (Biolgend, 358512), and CXCR5 (Biolgend, 145511) for the T cell staining panel, and CD19 (Biolgend, 152404), B220 (Biolgend, 103212), and CD138 (Biolgend, 142504) for the B cell staining panel. CD4 (Biolgend, 100204), CD25 (Biolgend, 113704), FOXP3 (Biolgend, 320014) for the Treg cell staining panel. Each of these antibodies was added to the appropriate sample tube, which was then incubated for 20 min. After incubation, cells were washed with staining buffer, centrifuged at 350 g for 5 min, and the supernatant was removed. Finally, cells were resuspended in 250 µL of cell staining buffer and analyzed using the Beckman CytoFLEX S flow cytometer.
Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of anti-dsDNA antibodies and ANA were measured using enzyme-linked immunosorbent assay. The assays were performed according to the manufacturer’s instructions using CUSABIO ELISA Kits, which employ a double antigen sandwich ELISA method. Additionally, TNF-α and IL-12p70 levels were determined using Multisciences ELISA Kits.
Renal Histology and Immunofluorescence
Mouse kidney tissues were fixed in a formaldehyde solution for 2 days, followed by dehydration, paraffin embedding, and sectioning. The sections were treated with HD constant staining pretreatment solution, stained with hematoxylin, and then rinsed under running water. Subsequently, sections were immersed in eosin dye. After dehydration and mounting, images were acquired and analyzed.
For immunofluorescence staining, the procedure was carried out at room temperature. Paraffin-embedded kidney sections were deparaffinized and rehydrated, followed by antigen retrieval. Hydrogen peroxide was used to block endogenous peroxidase activity, and a hydrophobic barrier was drawn around the tissue using a histochemical pen. The tissue was then blocked with serum and incubated with the primary C3 antibody (1:500, Thermo PA1-29715). Subsequently, the corresponding HRP-labeled secondary antibody (1:500, Servicebio GB23303) was applied, followed by the addition of the appropriate TSA dye. The tissue sections were placed in an antigen retrieval buffer for additional antigen retrieval steps. The IgG antibody (1:2000, Proteintech, 30000-0-AP) was then introduced, followed by the corresponding secondary antibody (1:500, Servicebio GB21303). The cell nuclei were counterstained with DAPI to minimize autofluorescence, and the sections were sealed. Finally, images were captured for analysis.
Transcriptome Analysis
Total RNA was extracted from spleen or kidney tissues using TRIzol® Reagent, following the manufacturer’s instructions. RNA quality was assessed using the 5300 Bioanalyzer (Agilent), and quantification was carried out with the ND-2000 spectrophotometer (NanoDrop Technologies). RNA purification, reverse transcription, library construction, and sequencing were conducted by Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China), according to the manufacturer’s protocols (Illumina, San Diego, CA) as previously described [28]. The raw sequencing reads have been deposited in the NCBI Sequence Read Archive (SRA) under Accession Number PRJNA1165911.
The raw paired-end reads were processed using fastp [29] for trimming and quality control, employing default parameters. Clean reads were then aligned to the reference genome using HISAT2 [30] with orientation mode. The mapped reads for each sample were assembled using StringTie [31] based on a reference-guided approach. Differential expression genes (DEGs) between different samples were identified by calculating the expression level of each transcript using the transcripts per million reads (TPM) method. Gene abundance quantification was performed with RSEM [32]. Differential expression analysis was conducted using DESeq2 [33], with criteria set to|log2FC| ≥ 2 and a false discovery rate (FDR) ≤ 0.05. Furthermore, KEGG pathway enrichment analysis was carried out to identify DEGs significantly enriched in metabolic pathways, using KOBAS [34] with a Bonferroni-corrected P-value ≤ 0.05, compared against the whole-transcriptome background. To clarify the expression of interferon (IFN) genes, we analyzed changes in IFN gene expression in the spleen and kidney after exposure to nitrite using transcriptomic data.
Western Blot Analysis
The expression of Itgam and Cadm1 was also assessed via Western blot analysis. Kindey samples (50 mg) were homogenized in 9 volumes (v/w) of RIPA buffer (Beyotime, P0013B) supplemented with protease inhibitor (Beyotime, P1005) to extract total protein. The total protein concentration was determined using the BCA protein assay kit (Beyotime, P0010S). Forty micrograms of protein from each sample were loaded onto a 10% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% skim milk powder in Tris-buffered saline containing 0.1% Tween-20 for one hour at room temperature, followed by overnight incubation with diluted primary antibodies against GAPDH (1:2000, Cell Signaling Technology, 5174 S), Itgam (1:1000, ABclonal A24120), or Cadm1 (1:1000, Thermo PA5-24196) at 4 °C. After incubation with the appropriate secondary antibody (1:10000, LI-COR, C50331-05) for 2 h at room temperature, the bands were scanned using an infrared imaging system (Odyssey, LI-COR) and quantified using ImageJ software.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism 8.0. Each experiment included a minimum of three biological replicates. Data were presented as mean ± standard deviation (± s). Parametric statistical methods were used for normally distributed data, while non-parametric tests were applied when the data did not meet this criterion. For comparisons across multiple groups, one-way analysis of variance (ANOVA) was employed. A p-value of < 0.05 was considered to indicate statistical significance.

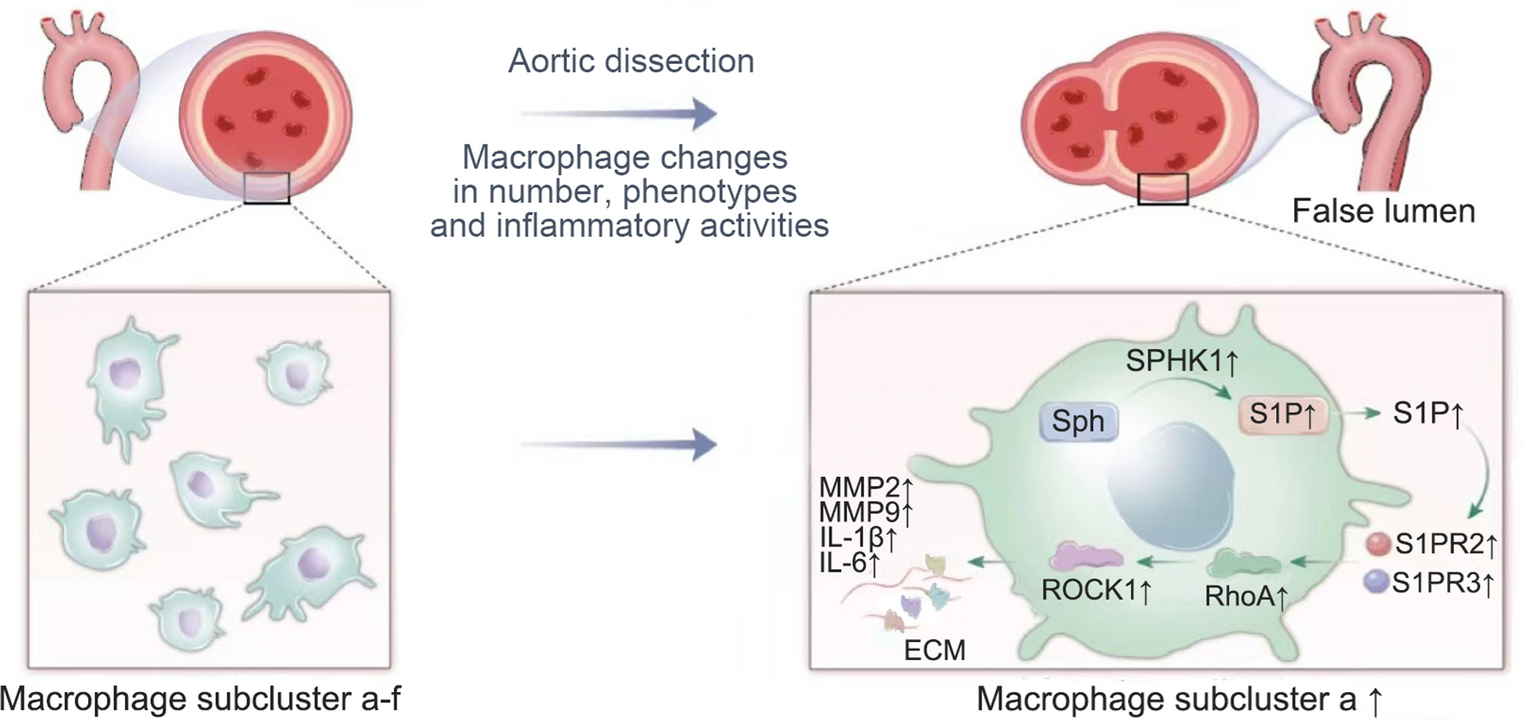
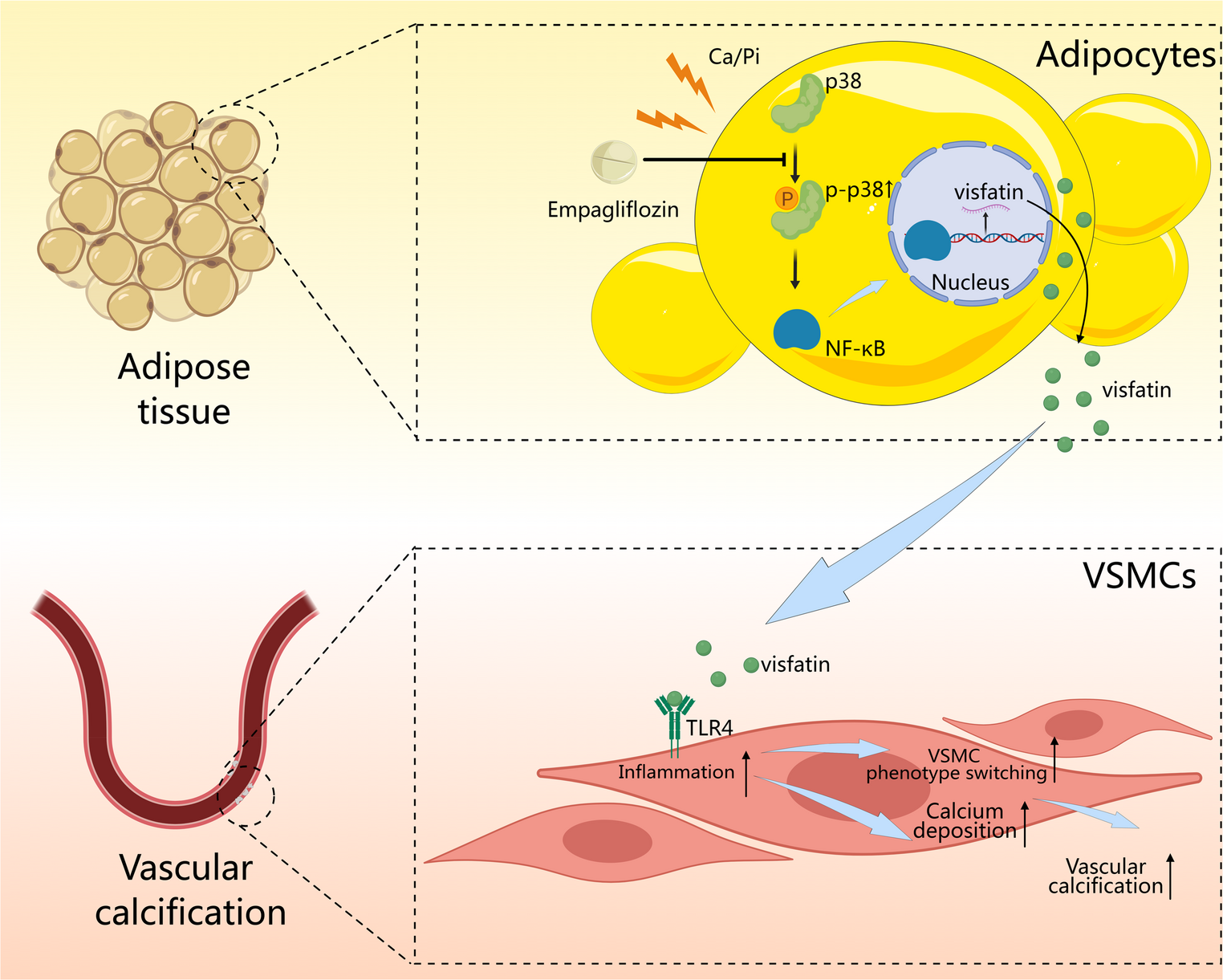
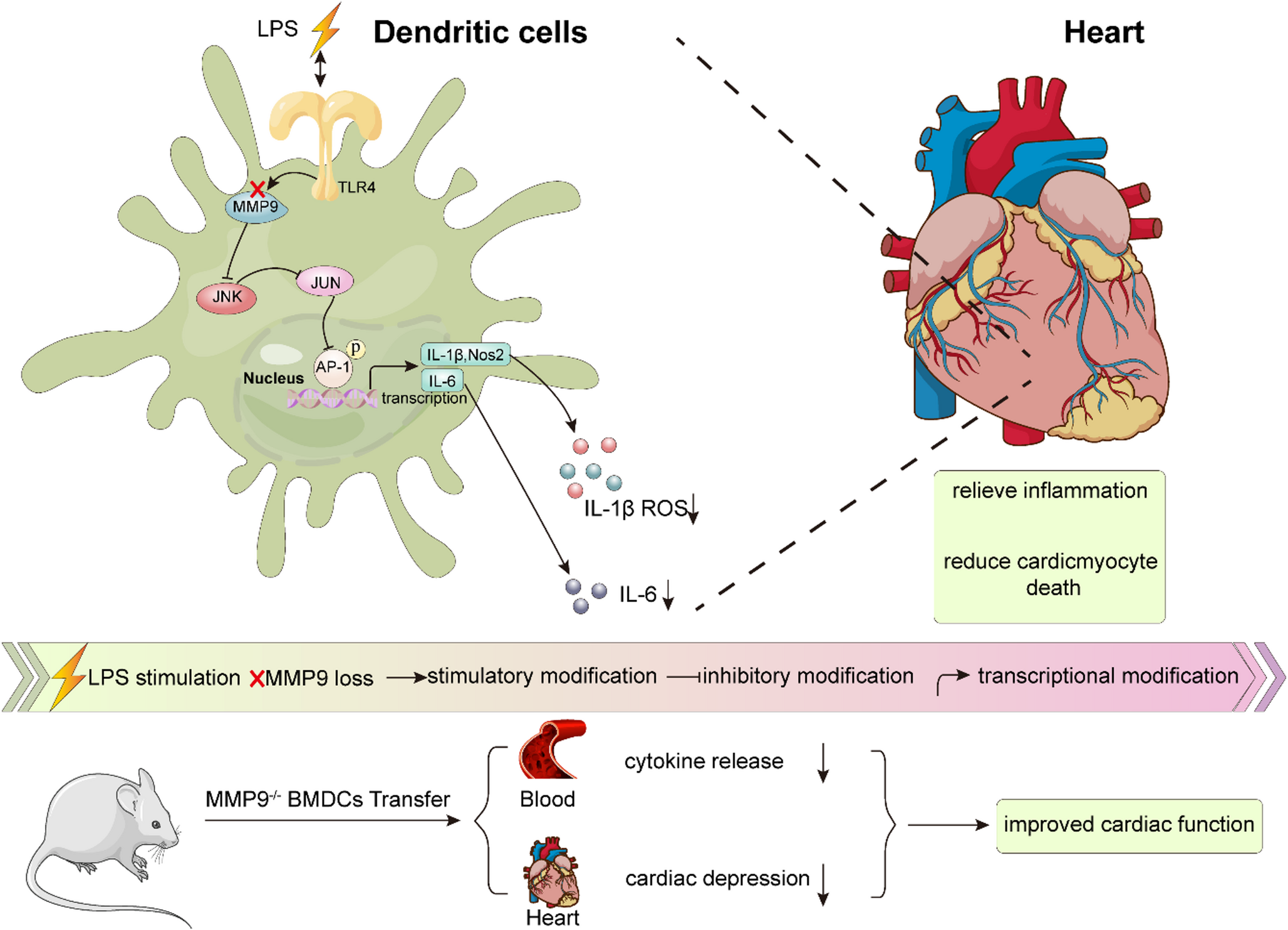
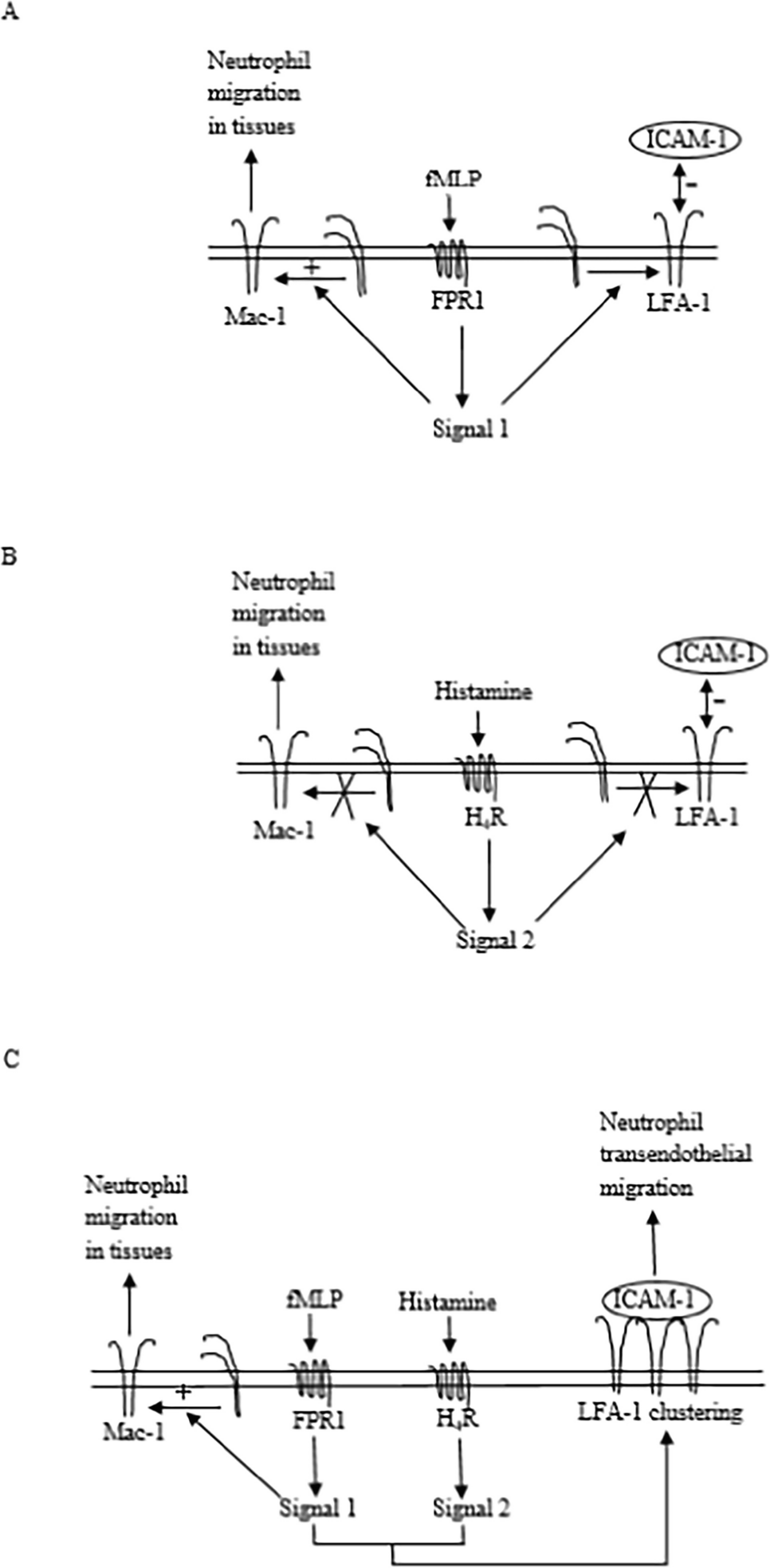


Comments (0)