
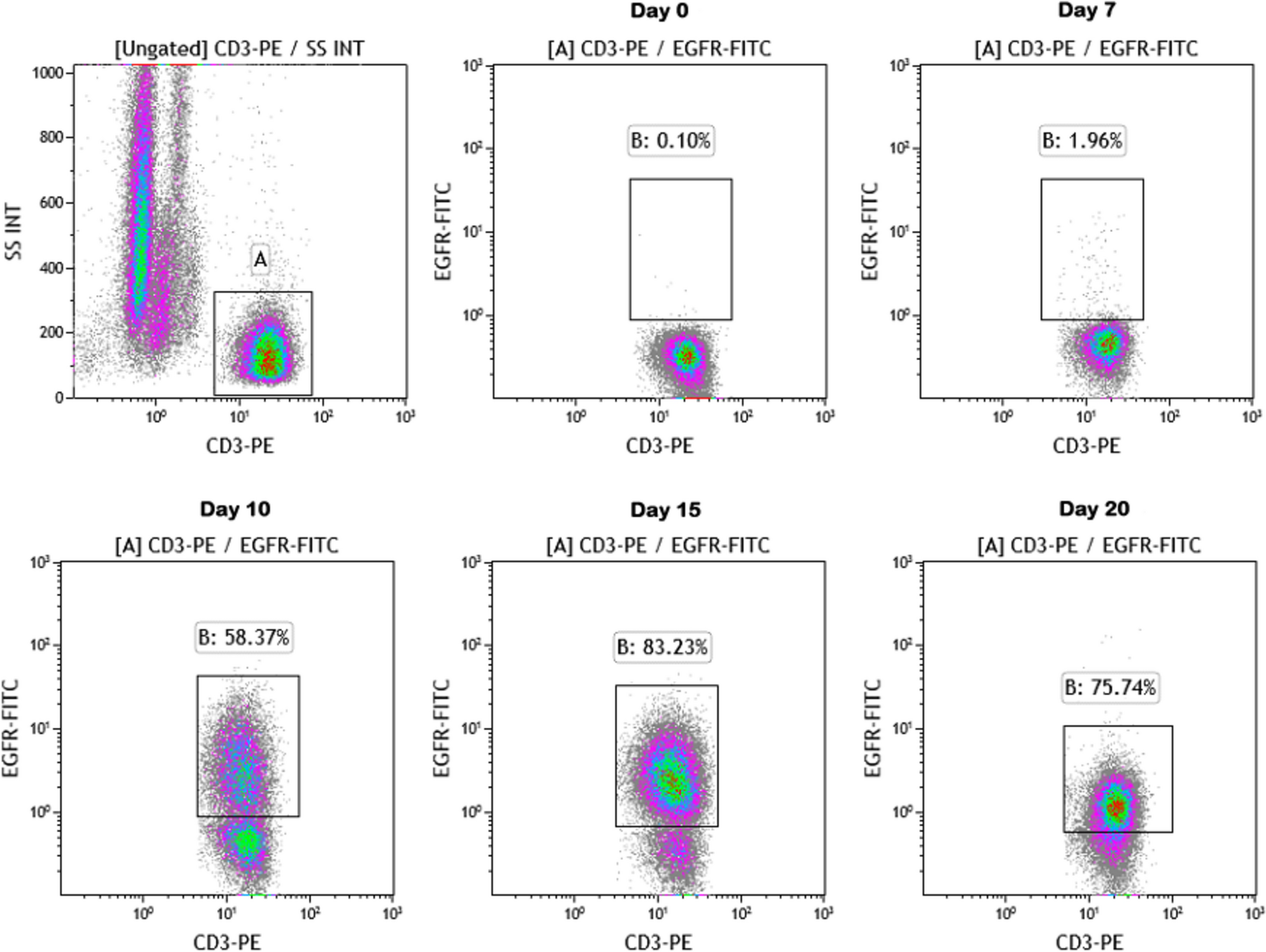
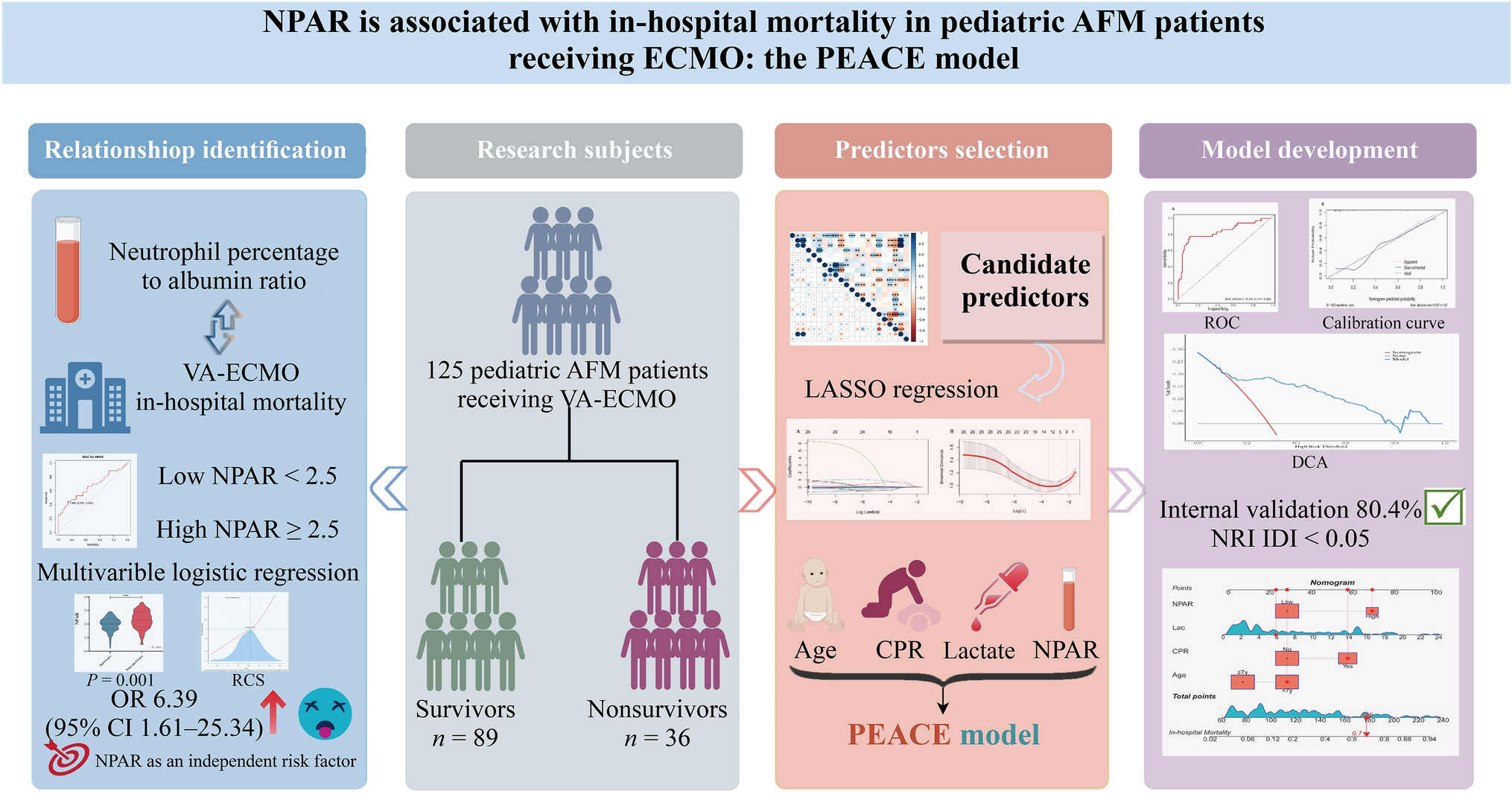
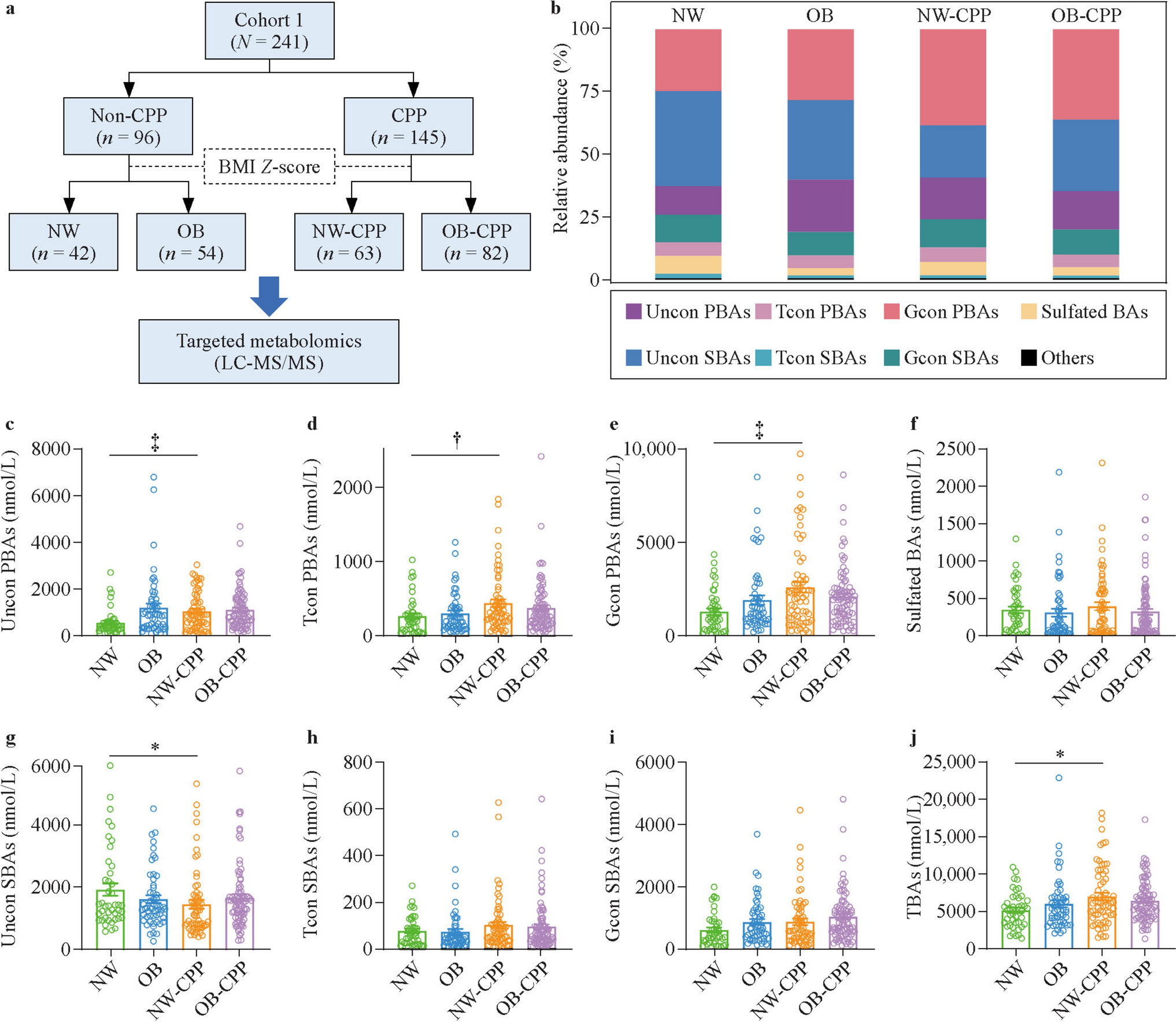
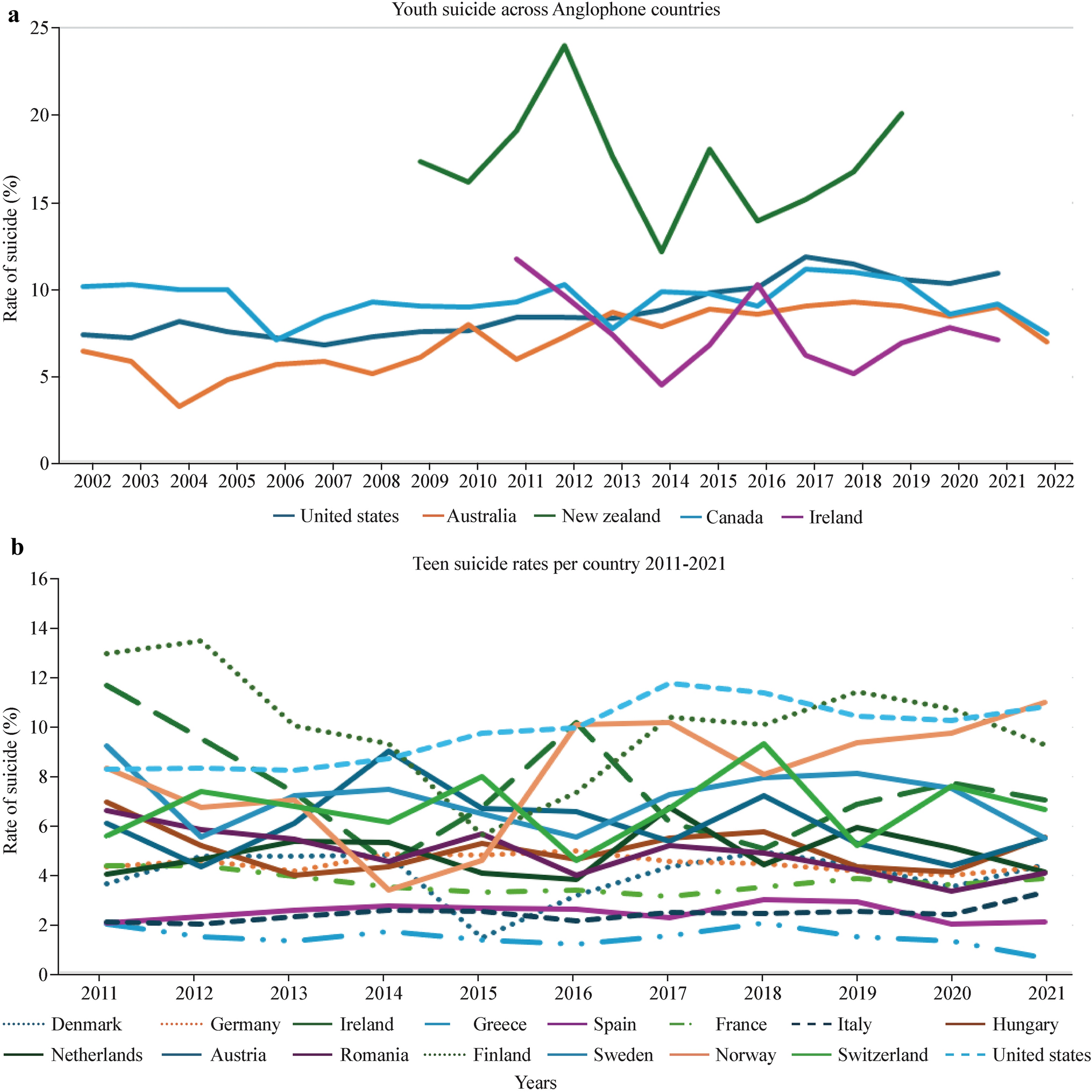
Remember me
The intersection between obesity-related metabolic inflammation and hypothalamic–pituitary–gonadal (HPG) axis dysfunction in adolescents has emerged as one of the most significant paradigm shifts in contemporary reproductive endocrinology [1]. Mounting evidence indicates that chronic low-grade inflammation induces both functional and structural changes in the hypothalamus, resulting in reproductive dysfunction that often precedes a formal diagnosis of polycystic ovary syndrome (PCOS) by several years [2]. This insidious progression, driven by multifaceted mechanisms including gliosis, endoplasmic reticulum (ER) stress, and peripheral immune cell chemotaxis, underscores the inadequacy of current diagnostic paradigms—which rely on late-stage phenotypic manifestations—for identifying at-risk youth.
The pathophysiological triad originally described (1) cytokine-mediated leptin resistance in kisspeptin neurons [3], (2) increased blood–brain barrier permeability [4], and (3) gut dysbiosis-driven systemic inflammation [5] has currently been expanded to include gliosis, endoplasmic reticulum stress, and vascular adaptations [3,4,5] (Fig. 1). These interconnected processes, together with peripheral immune cell infiltration [4] and gut-derived inflammatory mediators [5], create a neuroinflammatory milieu that disrupts gonadotropin-releasing hormone (GnRH) pulsatility [3], ultimately manifesting as detectable ovarian stromal changes [6]. Critically, gliosis amplifies cytokine signaling through microglial activation [4], whereas ER stress impairs roopiomelanocortin (POMC) and kisspeptin neuronal function [3, 5], which are mechanisms that synergize with leptin resistance to perpetuate reproductive dysfunction [3, 5]. The temporal progression from hypothalamic inflammation to end-organ pathology [6] underscores the need for time-sensitive interventions [4].
Fig. 1
Hypothalamic inflammation and leptin resistance disrupt the hypothalamic–pituitary–gonadal (HPG) axis in obesity. Chronic low-grade inflammation, secondary to obesity and gut dysbiosis, disrupts the hypothalamic–pituitary–gonadal (HPG) axis. Elevated levels of TNF-α and IL-6 enter the brain through increased blood–brain barrier permeability and gliosis, inducing endoplasmic reticulum stress in the arcuate nucleus, which leads to central leptin resistance. Despite high circulating leptin levels, kisspeptin expression is impaired. This reduces GnRH release and subsequently decreases LH and FSH secretion from the pituitary gland. These alterations are often associated with oligomenorrhea or polycystic ovary syndrome-like phenotypes that may not meet full diagnostic criteria. Additionally, this neuroendocrine disruption contributes to a vicious cycle of increased food intake, reduced energy expenditure, worsened insulin resistance, and further weight gain. ArcN arcuate nucleus, BBB blood–brain barrier, FSH follicle-stimulating hormone, GnRH gonadotropin-releasing hormone, IL-6 interleukin-6, LH luteinizing hormone, LR leptin resistance, MSH melanocyte-stimulating hormone, POA preoptic area, POMC pro-opiomelanocortin, PVN paraventricular nucleus, TNF-α tumor necrosis factor alpha
Recent findings indicated that 68% of adolescents with obesity exhibit HPG axis disruption detectable through steroid metabolomics, even in the presence of normal conventional laboratory parameters, bringing several important clinical implications. First, these data highlight that standard endocrine assays [e.g., serum luteinizing hormone (LH), follicle-stimulating hormone (FSH), testosterone, and estradiol] may lack sensitivity for detecting early or subtle HPG axis dysfunction in this population. Steroid metabolomic profiling, as demonstrated by Gawlik et al., can reveal distinct biochemical signatures of HPG axis perturbation—such as altered excretion of C19 and C21 steroids, evidence of partial 21-hydroxylase or 3β-hydroxysteroid dehydrogenase insufficiency, and imbalances in 11β-hydroxysteroid dehydrogenase activity—well before overt clinical or biochemical hypogonadism is evident by conventional testing [7].
This subclinical disruption is clinically significant, because it may precede the development of overt reproductive or metabolic pathologies, including PCOS, hypogonadism, and associated comorbidities such as insulin resistance, hypertension, and hepatic steatosis [8]. The high prevalence of these metabolomic abnormalities suggests that HPG axis dysfunction is a common, early, and potentially modifiable consequence of pediatric obesity, rather than a late-stage complication. Furthermore, the heterogeneity of steroid metabolomic signatures among obese adolescents indicates that obesity-related HPG axis dysfunction is not a monolithic entity, but may reflect distinct pathophysiological subtypes, each with unique risk profiles and therapeutic implications [7].
We propose the formal recognition of this entity as metabolic inflammatory hypothalamic-induced reproductive dysfunction (MIHRD) to encapsulate the early obesity-related disruption of neuroendocrine control within the HPG axis. This nomenclature highlights the central role of neuroinflammation and its reproductive-specific consequences, positioning MIHRD as a pathophysiological continuum rather than a static condition. Recognizing MIHRD may prompt clinicians to implement earlier, targeted interventions in high-risk youth—potentially altering disease trajectories before irreversible damage occurs [9].
Although formal diagnostic criteria for the MIHRD have yet to be established, the synthesis of current evidence supports its conceptual validity. Future research should prioritize the development and validation of such criteria, potentially incorporating novel tools such as steroid metabolomics and neuroimaging markers of hypothalamic inflammation. These steps will be essential to refine clinical risk stratification and optimize therapeutic timing.
Future interventions should target not only systemic inflammation but also hypothalamic-specific processes (e.g., microglial activation and ER stress relief) to restore reproductive function. Emerging data also suggest that the modulation of neuroinflammation through lifestyle and pharmacological strategies—such as anti-inflammatory dietary patterns, physical activity, metformin, and glucagon-like peptide-1 (GLP-1) receptor agonists—may offer therapeutic benefits by restoring hypothalamic homeostasis and improving both reproductive and metabolic outcomes [10, 11]. Therefore, future investigations should focus not only on refining diagnostic biomarkers but also on evaluating whether early reversal of hypothalamic inflammation can prevent the downstream cascade of PCOS and metabolic syndrome, particularly in genetically or epigenetically predisposed individuals.
In conclusion, obesity-induced hypothalamic inflammation represents a foundational and underrecognized driver of adolescent reproductive dysfunction. The concept of the MIHRD provides a unifying framework for understanding the early neuroendocrine alterations that precede overt PCOS or hypogonadism. Integrating advanced diagnostic tools such as steroid metabolomics with targeted neuroimmune interventions may redefine how we identify and manage at-risk adolescents—ultimately shifting the paradigm from reactive treatment to proactive prevention.
Comments (0)