Epilepsy is a common chronic neurological disorder that affects people of all ages, ethnicities. The prevalence of epilepsy varies significantly between countries, being usually higher in low- and middle-income countries than in high-income countries (Walter & Ron 2011). The pathogenesis of epilepsy is complex, and the study of its mechanisms and pathological changes is crucial for the discovery of effective diagnostic and therapeutic targets.
ERS is a cellular response to the accumulation of unfolded or misfolded proteins within the ER lumen. When genetic or environmental factors overwhelm the protein-folding capacity of the ER, disruption of calcium homeostasis—caused by cytosolic Ca2⁺ overload and ER Ca2⁺ depletion—leads to the accumulation of unfolded proteins. This, in turn, activates the UPR to restore ER homeostasis (Xie et al. 2023). During epileptic seizures, abnormal neuronal discharges lead to a significant increase in intracellular Ca2⁺ levels, thereby inducing ERS. Additionally, the excessive generation of ROS during seizure activity triggers oxidative stress, which can directly damage molecular chaperones within the ER, resulting in the accumulation of misfolded proteins and further exacerbating ERS (Wang et al. 2023). Moreover, ATP depletion and energy failure caused by seizures may aggravate ERS, forming a vicious cycle. ERS, in turn, can activate pro-inflammatory pathways such as NF-κB, promoting the release of inflammatory cytokines from glial cells, which worsens seizure severity and neuronal injury (Sarawi et al. 2025). ERS also contributes to neuronal apoptosis by activating apoptotic signaling pathways. Studies have shown that the expression of CHOP and caspase-12 is significantly up-regulated in the hippocampus of epileptic mice, suggesting that ERS-mediated apoptosis may be involved in seizure-related neuronal loss (Doganyigit et al. 2023). Furthermore, ERS may influence seizure activity through the regulation of autophagy (Xie et al. 2020). Therefore, ERS represents a critical mechanism in the pathophysiology of epilepsy, and targeting ERS-related pathways may offer novel therapeutic strategies for epilepsy management.
The role of ERS in neurological disorders has increasingly become a research focus. In recent years, abnormal ERS has been observed in both human epilepsy and experimental epilepsy models (Yue et al. 2020). Moreover, interventions targeting ERS have demonstrated antiepileptic and neuroprotective effects in epilepsy (Qiao et al. 2024). These findings suggest that ERS holds great potential as a therapeutic target for epilepsy. Based on the multifaceted involvement of ERS in epilepsy, we performed bioinformatic analyses to identify ERS-related DEGs, followed by experimental validation to explore their functions and diagnostic value. Our aim is to discover novel therapeutic targets and provide new strategies for the diagnosis and treatment of epilepsy.
In this study, a bioinformatics-based framework was established to investigate the specific role of ERS in epilepsy from a holistic perspective, and 83 ERS-related DEGs in epilepsy were identified. BP analysis of ERS-related DEGs in epilepsy revealed that ‘apoptosis-regulating signaling pathway’ was the most significantly enriched pathway, suggesting that ERS plays an important role in regulating apoptosis in epilepsy. Studies have shown that neuronal apoptosis is one of the major pathological manifestations caused by epileptic seizures, which may lead to epileptogenesis and cognitive impairment (Yamamoto et al. 2006), and that chronic epilepsy is associated with ERS-induced pro-apoptotic and anti-apoptotic signaling pathways (Keestra-Gounder et al. 2016). Other processes that are enriched include the response to unfolded proteins, the ERS response, and the regulation of calcium homeostasis. These processes are commonly associated with cellular stress responses and protein folding, which is particularly important in the context of ERS. The enrichment for ‘response to hypoxia’ suggests that ERS is associated with cellular adaptive responses under hypoxic conditions, a common phenomenon in epilepsy (Xu & Fan 2022). CC analysis revealed that ‘endocytosis vesicles’ and ‘phagocytosis vesicles’ were the most enriched cellular components, suggesting that these vesicles may play a role in the cellular response to ERS in epilepsy, and may be associated with the transport and degradation of unfolded proteins or damaged cellular components. The enrichment of ER-related components, such as ‘integrative components of the endoplasmic reticulum membrane’ and ‘endoplasmic reticulum chaperone complexes,’ further emphasizes the central role of the ER in the stress response in epilepsy. MF analyses yielded a significant enrichment for ‘peptide binding,’ suggesting that many of the differential genes are involved in peptide binding processes (which may be associated with unfolded protein reactions or proteasomal degradation). Binding activities associated with protein ligases and proteases (e.g., ‘ubiquitin-like protein ligase binding’ and ‘protease binding’) highlight the role of protein homeostasis mechanisms in managing ERS reactions.
KEGG analysis revealed that ‘apoptosis’ was the most enriched KEGG pathway, suggesting that ERS-related DEGs in epilepsy play an important role in apoptotic signaling. This is consistent with the enrichment of apoptotic signaling mentioned in the BP analysis. Other notable pathways include ‘NOD-like receptor signaling pathway,’ ‘NF-κB signaling pathway,’ and ‘PI3K-Akt signaling pathway,’ which are associated with inflammatory response and stress response pathways, and are commonly associated with neurological damage and degeneration in epilepsy. The NOD-like receptor signaling pathway mediates ERS-triggered inflammatory response through NOD-1 and NOD-2, activates Receptor-interacting protein 2 and NF-κB pathways, and promotes inflammatory cytokine secretion (An et al. 2019; Wei et al. 2022). NLRP3 inflammatory vesicles interact with ERS in epilepsy (Yamamoto et al. 2006). Dual role of NF-κB in epilepsy: up-regulation in the short term is neuroprotective but can also trigger a deleterious inflammatory response (Cai & Lin 2022). The PI3K-Akt signaling pathway regulates the NF-κB pathway through activation of Akt proteins, promotes the secretion of inflammatory factors, and helps cells to maintain cellular homeostasis in response to oxidative stress and ERS (Tian et al. 2022). ‘Calcium signaling’ and ‘AMPK signaling’ were also shown to be enriched, highlighting the role of calcium homeostasis and energy metabolism in regulating cellular stress responses.
ERS-related DEGs in epilepsy are mainly involved in the regulation of apoptosis, protein folding, and cellular stress response, in which the ER plays an important role. In addition, signaling pathways related to inflammation, cell survival, and metabolic stress are widely activated, suggesting that ERS may be closely linked to cell injury and compensatory stress responses.
To further identify key targets with high diagnostic potential among ERS-related DEGs in epilepsy, we constructed a PPI network using the STRING database and identified eight Hub genes using Cytoscape. These Hub genes are CXCL8, TLR4, MMP9, TNFRSF1A, PTGS2, STAT1, BCL2, and RELA. They are closely associated with immune regulation, inflammatory responses, and apoptosis, processes that are considered potential mechanisms for the onset and progression of epilepsy.
CXCL8 is involved in various biological processes, including the promotion of inflammatory responses, regulation of immune function, and angiogenesis. During inflammation, CXCL8 recruits, activates, and induces the migration of neutrophils to the site of inflammation, and also exerts chemotactic effects on other immune cells such as monocytes and T cells, thereby participating in a broad spectrum of immune responses (Cambier etv al. 2023; Liba et al. 2019). CXCL8 may also contribute to the pathogenesis of epilepsy through immune regulatory mechanisms associated with ERS. Studies have shown that the UPR pathway can modulate inflammation via signaling pathways such as NF-κB, thereby promoting the secretion of CXCL8. Notably, the NF-κB pathway has been closely linked to the onset and progression of epilepsy (Hsu et al. 2024). Furthermore, the inflammatory cytokine CXCL8 can enhance cellular responses to ERS by activating signaling pathways including SHP2, ERK, and p38 MAPK (An et al. 2019; Ding & Wu 2024).
Studies have reported an upward trend in CXCL8 levels in both peripheral blood and cerebrospinal fluid of epilepsy patients (Howe et al. 2023; Mazdeh et al. 2018). However, in the present study, we found that CXCL8 expression in the peripheral blood of epilepsy patients was significantly lower than that of healthy controls, which contrasts with the previous findings. This discrepancy may be attributed to differences in patient populations. For instance, the study by Mehrdokht et al. analyzed blood samples from seizure-free individuals treated with valproate for at least six months, while Charles et al. focused on newly diagnosed patients with drug-resistant epilepsy. In contrast, the GEO dataset used in our study included data from drug-naïve patients or those treated with monotherapy who had experienced their most recent seizure within 72 h prior to sample collection. Another potential explanation is the difference in sampling time. Our dataset includes patients who had seizures within 72 h and newly diagnosed patients who had not yet received treatment. Therefore, the observed decrease in peripheral CXCL8 expression may reflect complex alterations in inflammation and immune responses in epilepsy patients. This hypothesis, however, requires further validation through longitudinal studies.
The ROC curve analysis for the hub gene CXCL8 revealed an AUC value of 0.78, indicating its potential as an auxiliary diagnostic biomarker. However, its standalone diagnostic performance is not sufficient to replace established gold standards such as electroencephalography (EEG). Future research may focus on developing combined diagnostic models incorporating CXCL8 with other ERS-related genes, such as TLR4 and STAT1, or integrating CXCL8 into existing clinical scoring systems to enhance diagnostic sensitivity and specificity. In conclusion, further investigation is warranted to better understand the expression pattern and diagnostic value of CXCL8.
TLR4 mainly mediates immune responses by recognizing lipopolysaccharide or bacterial endotoxins and is involved in innate immunity. Upon activation, TLR4 induces the transcription of cytokine genes and activates the NF-κB signaling pathway, promoting inflammation. This pathway may exacerbate neuronal damage and drive the pathological progression of epilepsy (Kang et al. 2023). The NF-κB pathway also plays a role in regulating immune responses following ERS activation (Ming et al. 2022). In this study, we found that TLR4 expression in the peripheral blood of epilepsy patients was elevated compared to healthy controls, possibly due to immune system activation triggered by epilepsy seizures. Once activated, TLR4 not only promotes immune cell migration but may also exert feedback effects on local immune responses through cytokine secretion and immune cell activation. Furthermore, TLR4 may influence the immune state of the brain by crossing the blood–brain barrier (BBB), enhancing neuroinflammatory responses and exacerbating epilepsy (Foiadelli et al. 2023). Recent research in animal models has shown that TLR4 is a key inflammatory trigger by inducing the transcription of several cytokine genes, promoting the occurrence and severity of epilepsy seizures (Maroso et al. 2010). Studies have also reported a strong correlation between TLR4 expression and the frequency of epilepsy seizures (Pernhorst et al. 2013), which is consistent with our findings of elevated TLR4 expression in the peripheral blood of epilepsy patients. In conclusion, we consider TLR4 to be a core immune regulatory factor that serves not only as a marker of immune responses but also as a key regulator of epilepsy seizures. Modulating TLR4 activity may provide new therapeutic strategies for the immunotherapy of epilepsy.
MMP9 is a matrix metalloproteinase involved in the degradation of the extracellular matrix, cell migration, and tissue remodeling. Its activity is closely related to ERS (Lin et al. 2020). Studies have shown that after ERS activation, MMP9 expression can increase, exacerbating neuroinflammation, particularly through the disruption of the BBB, which is a major factor in the onset of epilepsy and neuronal damage (Hu et al. 2024). MMP9 has been shown to contribute to the occurrence and development of epilepsy, with multiple studies demonstrating increased expression of MMP9 in the serum of epilepsy and epileptic seizure patients (Cai et al. 2020; Cudna et al. 2017; Ruber et al. 2018). In this study, MMP9 expression was elevated in the peripheral blood of epilepsy patients, reflecting an active state of neuroinflammation. The increase in MMP9 may degrade tight junction proteins in the BBB, leading to increased permeability and allowing immune cells and inflammatory factors to more easily enter the brain, thereby exacerbating local neuroinflammation and creating a vicious cycle (Yu et al. 2022). Overactivation of MMP9 leads to the degradation of the extracellular matrix, aggravating neuronal damage, while also enhancing immune cell migration and the secretion of inflammatory cytokines, further amplifying immune responses during epilepsy seizures (Gong et al. 2008; Regalado & Balogh 2024). In conclusion, we believe that the elevated expression of MMP9 may serve as a marker of the pathological process in epilepsy.
TNFRSF1A is a tumor necrosis factor receptor that binds to TNF-α, activating the MAPK and NF-κB signaling pathways. It plays a critical role in inflammatory diseases (Jing et al. 2022). The NF-κB pathway also plays a role in regulating immune responses following ERS activation. Additionally, TNFRSF1A participates in the activation of apoptosis. In this study, we found that TNFRSF1A expression was increased in the peripheral blood of epilepsy patients, suggesting that the immune system may be overactivated after the onset of epilepsy, potentially exacerbating neuroinflammation and neuronal damage, particularly under the activation of the TNF-α/TNFRSF1A signaling pathway (Xu et al. 2020a, b). Peripheral immune cells may also migrate to the CNS, further aggravating neuronal damage and accelerating epilepsy seizures. Therefore, the up-regulation of TNFRSF1A can serve as an indicator of intense inflammatory responses and provides new targets for the diagnosis and treatment of epilepsy.
PTGS2, commonly known as COX-2, is an important enzyme in the inflammatory response, involved in the synthesis of prostaglandins and the regulation of inflammation and pain. PTGS2 is associated with ERS-mediated neuroinflammation, and studies suggest that after epilepsy seizures, PTGS2 expression increases significantly, potentially leading to secondary brain damage and increasing the likelihood of recurrent seizures. PTGS2 plays an important role in the inflammation and hyperexcitability of the brain following seizures (Rojas et al. 2014). In the brains of epileptic rats, inhibition of NF-κB also suppressed PTGS2 expression (Xu et al. 2020a, b). In this study, we observed a significant decrease in PTGS2 expression in the peripheral blood of epilepsy patients. This phenomenon may be the result of systemic immune feedback, where the immune system prevents excessive activation to avoid neuronal damage during long-term seizures. This negative feedback may lead to a reduction in PTGS2 expression. Additionally, during epilepsy, PTGS2 expression is primarily concentrated in the brain’s damaged regions. Due to immune system regulation, the down-regulation of PTGS2 in peripheral blood does not indicate a weakened immune response but reflects the differences in central and peripheral inflammatory reactions. Therefore, low expression of PTGS2 in peripheral blood may serve as an important indicator for assessing the immune status and treatment response of epilepsy patients.
STAT1 is a nuclear transcription factor that regulates genes related to the cell cycle, cell survival, and immune response (Butturini et al. 2020). It plays a crucial role in the immune response as it is stimulated by interferons (IFNs) and inflammatory cytokines, thereby regulating the activation of immune cells and the release of cytokines (Liu et al. 2024a, 2024b). Studies have shown that the activation of STAT1 is closely related to ERS, especially during neuroinflammation and apoptosis. By activating related cytokines and gene expression, STAT1 further exacerbates the immune response (Li et al. 2019). The activation of STAT1 may enhance neuroinflammatory responses and apoptosis in epilepsy, promoting the pathological progression of epilepsy. In this study, the increased expression of STAT1 in the peripheral blood of epilepsy patients may represent excessive activation of the immune system. STAT1 activation promotes the migration of immune cells, which may lead to inflammatory cells entering the epileptic focus, further exacerbating neuronal damage. Additionally, during the onset of epilepsy, ERS activates STAT1, thereby enhancing immune response and apoptosis. This plays an important role in epileptic seizures and neuronal damage. Therefore, changes in STAT1 expression may serve as a useful marker for disease activity in epilepsy.
BCL2 is an important anti-apoptotic protein that plays a key role in cell survival (Moriishi et al. 2011). ERS typically regulates BCL2 through inositol-requiring enzyme 1 and CHOP, modulating its expression under cellular stress. During epileptic seizures, BCL2 helps protect neurons by reducing neuronal apoptosis. However, in the case of ERS activation, BCL2 may be inhibited through CHOP up-regulation, leading to increased neuronal apoptosis and exacerbating neuroinflammation in epilepsy (Fan et al. 2024). In this study, the down-regulation of BCL2 expression in the peripheral blood of epilepsy patients may be related to neuroinflammatory responses. After an epileptic seizure, inflammatory cytokines activate the NF-κB pathway, which in turn suppresses BCL2 expression and promotes neuronal cell apoptosis. Additionally, ERS activation also inhibits BCL2 expression. In the chronic progression of epilepsy, the down-regulation of BCL2 may reflect the immune system’s negative feedback mechanism, whereby the immune system modulates the immune response and reduces neuronal damage by inhibiting the expression of anti-apoptotic proteins under conditions of excessive neuronal activation and immune cell overreaction.
RELA encodes the p65 protein, one of the five transcription factors in the NF-κB family. The NF-κB pathway plays a key role in mediating inflammation and regulating immune responses, determining immune cell proliferation, apoptosis, and inflammation (An et al. 2023). After ERS activation, the RELA pathway is up-regulated, activating NF-κB, which enhances immune response and inflammation. This process plays an important role in the onset of epilepsy (Ming et al. 2022). In this study, RELA expression was found to be significantly elevated in the peripheral blood of epilepsy patients, suggesting enhanced immune response and exacerbated inflammation. During epileptic seizures, the NF-κB signaling pathway is induced by inflammatory cytokines and immune activation, leading to RELA activation. Meanwhile, the immune response in the nervous system triggers the release of cytokines such as TNF-α, IL-1β, and IL-6, which further enhance the immune response through the NF-κB pathway activated by RELA(An et al. 2019), promoting neuronal damage. RELA is also involved in the regulation of apoptosis, a process that exacerbates the persistence of epileptic seizures and neuronal damage.
In summary, the expression products of the identified hub genes play critical roles in both epilepsy and ERS, with mechanisms closely associated with inflammatory responses, immune regulation, and apoptosis—findings that are well aligned with the KEGG pathway enrichment analysis. However, this study is based on the analysis of peripheral blood mRNA, which does not directly reflect ERS activity within the brain. Additionally, the cross-sectional design limits the ability to establish a causal relationship between ERS-related gene expression and seizure activity. Therefore, future translational research will require validation in larger, well-characterized cohorts. Moreover, the specific mechanisms by which these hub genes influence epilepsy require further in-depth, multi-dimensional investigation.
Additionally, while this study offers valuable insights into the potential role of ERS in epilepsy, it has several limitations. First, epilepsy is a multifactorial disorder that cannot be fully explained by a single pathway. Our analysis focused exclusively on ERS-related genes, which limit the scope of interpretation. Second, although bioinformatics methods based on gene chip data have strong predictive capabilities, the study lacks experimental validation through cellular or animal models. While qRT-PCR confirmation of the most promising genes is valuable, it is insufficient for a comprehensive functional understanding. Future studies should integrate experimental models to further validate gene expression and investigate the functional roles of these differentially expressed genes in epilepsy. Additionally, the relatively small sample size may impact the generalizability and robustness of the findings. Although several ERS-related DEGs were identified through bioinformatic analysis, validation in larger clinical cohorts is crucial to determine their applicability across diverse populations. Finally, despite these limitations, the results of this study align with previous research on ERS and epilepsy, providing important insights and directions for future research in this area.



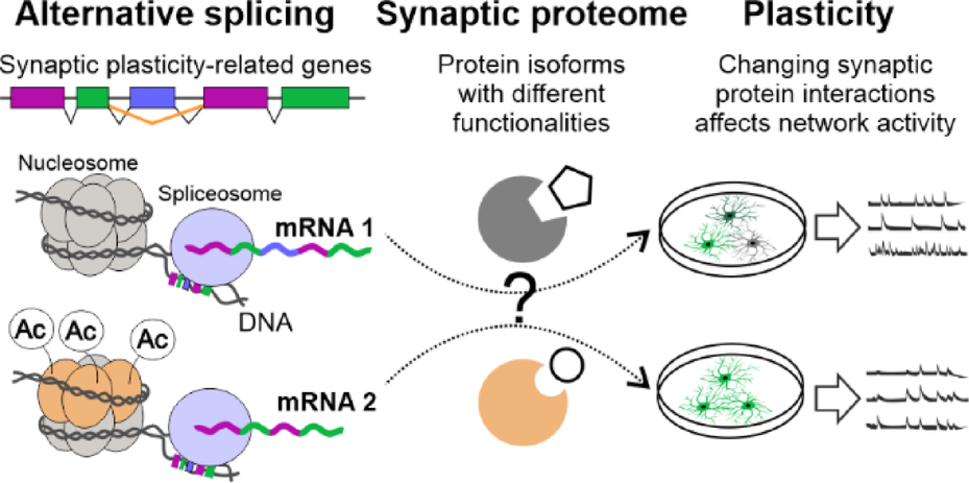
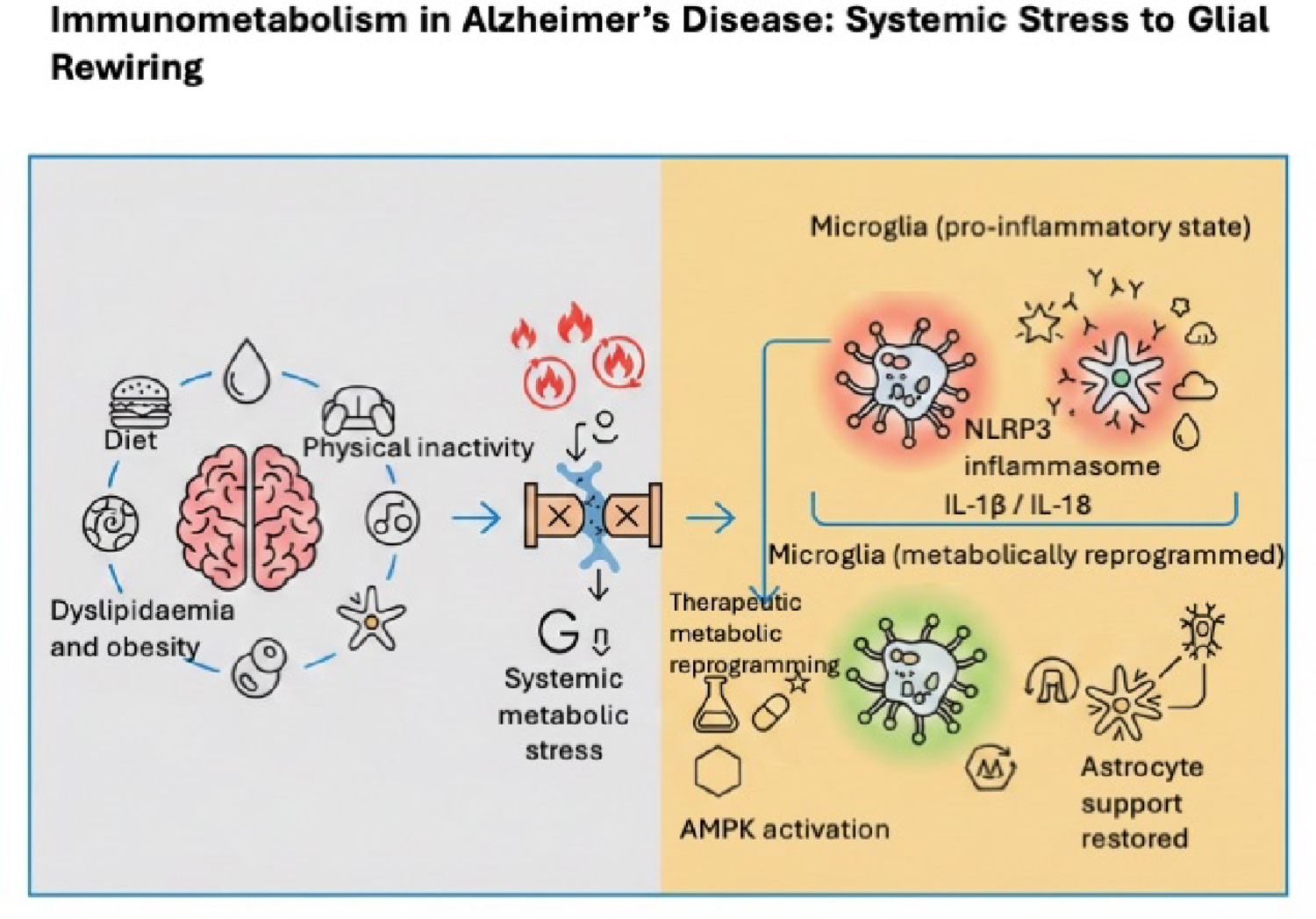
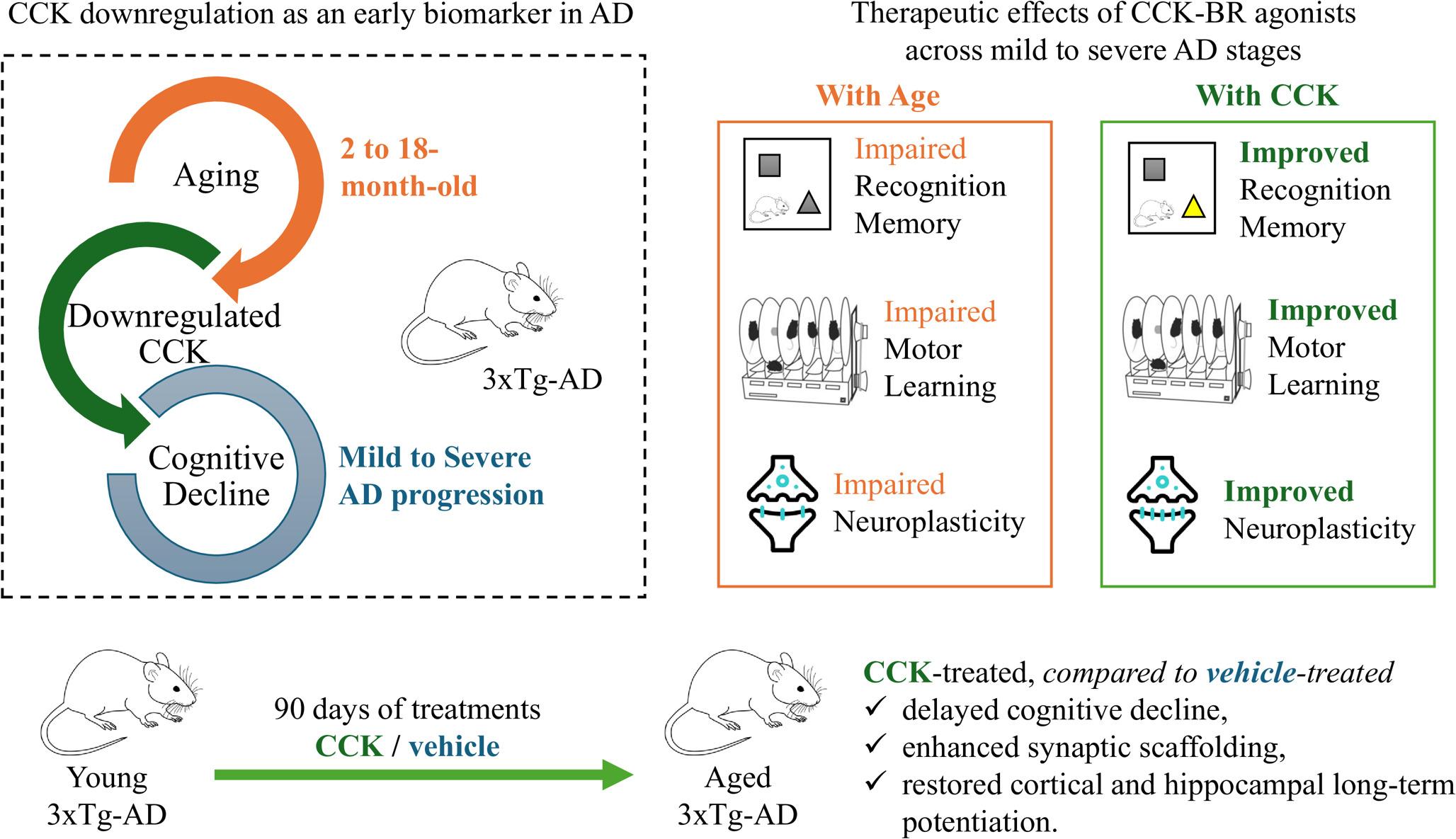
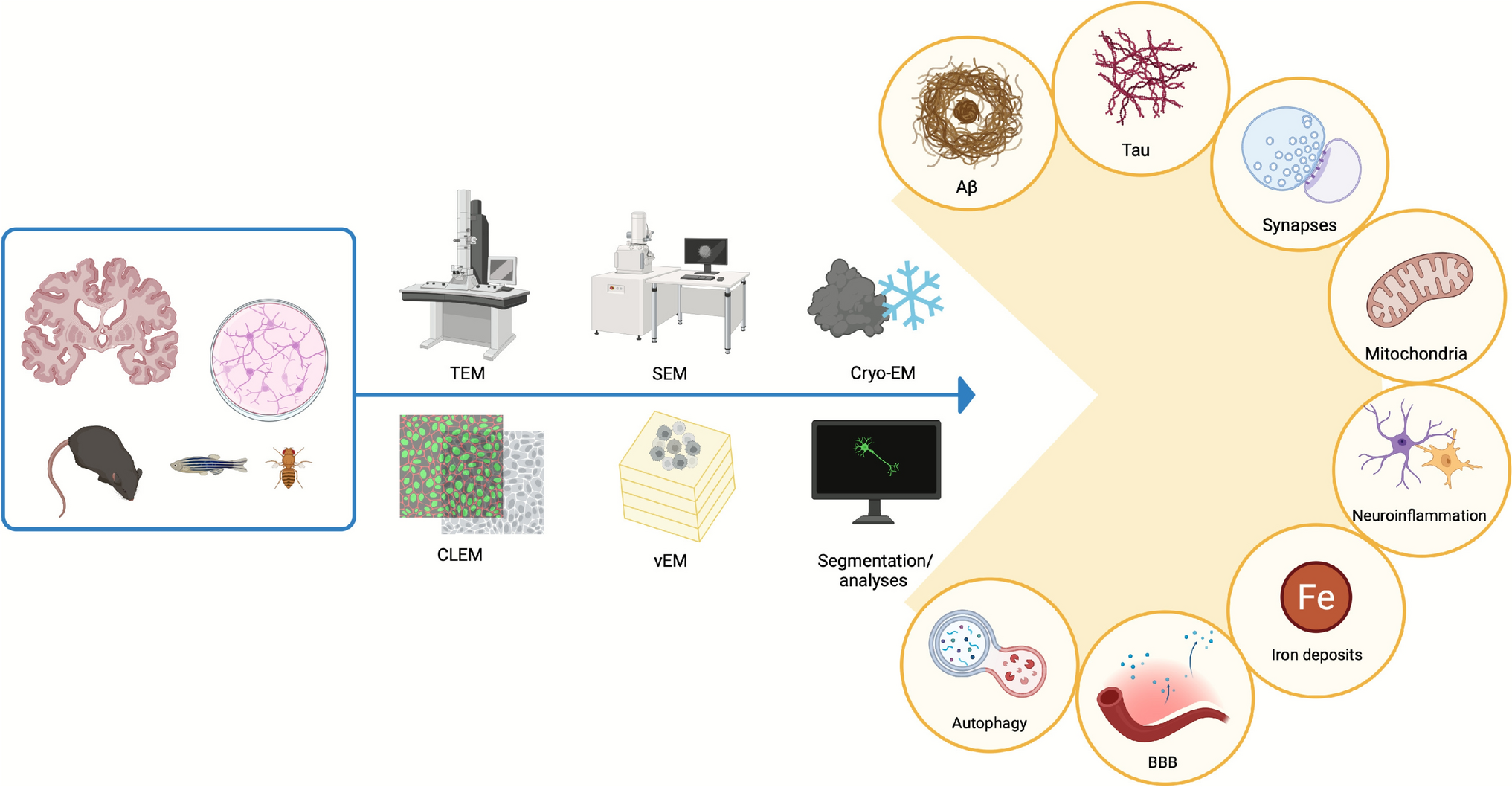
Comments (0)