
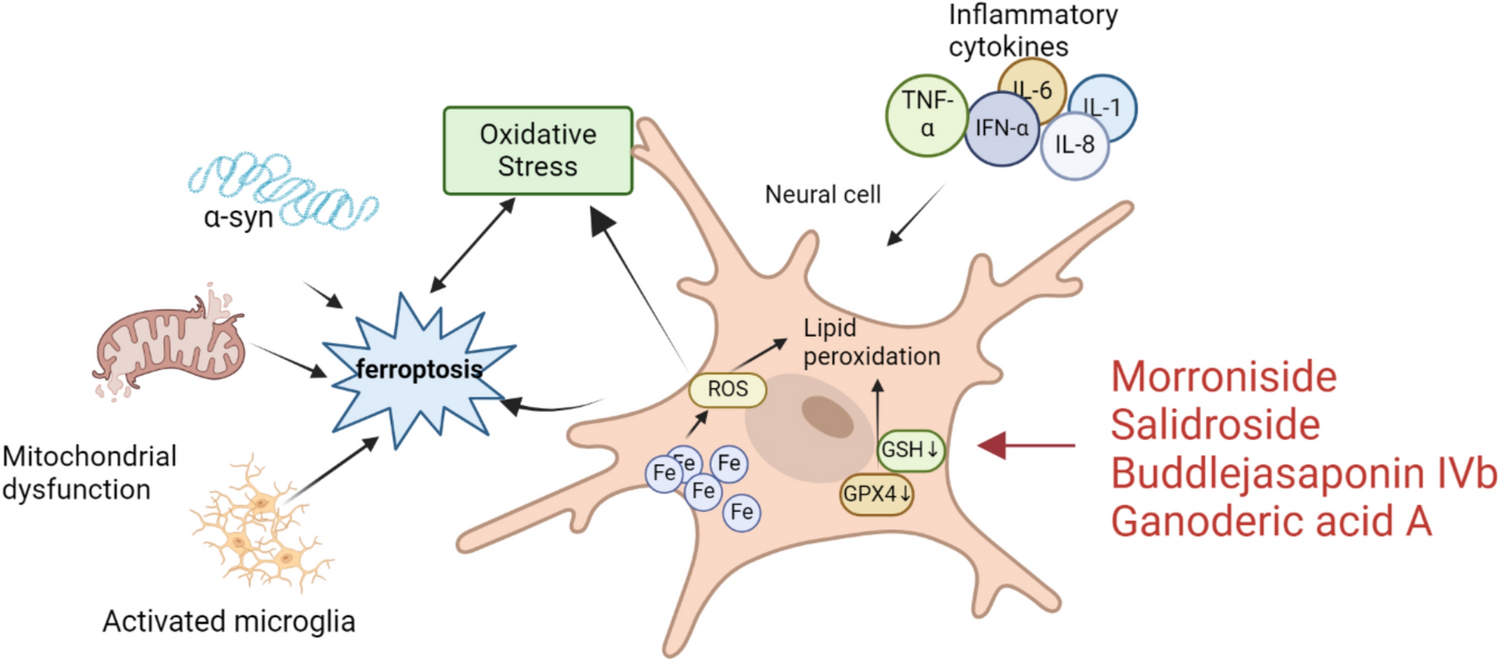
Remember me

We analyzed the GSE264648 expression profile containing 49 Alzheimer’s disease-related tissue samples. Initially, the Seurat package was used to process the data by reading the expression profiles and filtering cells based on several quality control metrics, including the number of unique molecular identifiers (UMIs) and the number of genes detected per cell, as well as the proportion of mitochondrial reads. Outlier cells were identified and excluded if they deviated by more than three median absolute deviations (MAD) from the median values. Specifically, cells with low gene expression (defined as nFeature_RNA > 200nFeature\_RNA > 200nFeature_RNA > 200, percent.mt ≤ 3 × MADpercent.mt \leq 3 \times MADpercent.mt ≤ 3 × MAD, nFeature_RNA ≤ 3 × MADnFeature\_RNA \leq 3 \times MADnFeature_RNA ≤ 3 × MAD, nCount_RNA ≤ 3 × MADnCount\_RNA \leq 3 \times MADnCount_RNA ≤ 3 × MAD, and percent.ribo ≤ 3 × MADpercent.ribo \leq 3 \times MADpercent.ribo ≤ 3 × MAD) were filtered out using violin plots and scatter plots for visual inspection.
Additionally, the DoubletFinder package was employed to detect and remove doublets, which are instances where two cells were mistakenly captured as a single unit. After filtering, 161,594 cells were retained for downstream analysis of feature expression levels, as shown in Supplementary Figs. 1A–1B. Supplementary Fig. 1C highlights the top 10 genes with the highest standard deviation (SD), which were selected to further explore cell heterogeneity in the dataset.
Cell Subpopulation Annotation and CommunicationBatch effects between the samples were first assessed using PCA dimensionality reduction analysis, as shown in Supplementary Fig. 1D. To mitigate these effects, further dimensionality reduction was performed using Harmony analysis, which successfully corrected for batch effects (Supplementary Fig. 1E). The optimal number of principal components (PCs) was determined by ElbowPlot, which indicated that 20 PCs should be used for further analysis (Supplementary Fig. 1F). UMAP (Uniform Manifold Approximation and Projection) analysis identified 16 distinct subtypes of cells (Fig. 1A). These subtypes were subsequently annotated into eight major cell categories: astrocytes, endothelial cells, PVMs, oligodendrocytes, oligodendrocyte precursor cells, vascular leptomeningeal cells, excitatory neurons, and inhibitory neurons (Fig. 1B). The bubble plots in Fig. 1C display the classic markers for each cell subtype. Additionally, Fig. 1D shows the relative proportions of each cell type in the case and control groups. As shown in Fig. 1D, distinct shifts in cell type proportions were observed under disease conditions. Specifically, the proportions of inhibitory neurons and excitatory neurons exhibited significant increases, whereas oligodendrocyte precursor cells (OPCs) and mature oligodendrocytes displayed marked reductions. In contrast, the proportion of PVMs showed no statistically significant alteration. These findings suggest that the pathological role of PVMs in disease is likely mediated by functional adaptations—such as enhanced cytokine secretion or modified cell–cell communication—rather than changes in proliferation or recruitment.
Fig. 1
Annotation of Cells A We utilized the principal component data from PCA and successfully divided the cells into 16 clusters using the UMAP algorithm. The formation of these clusters is based on the similarities and differences of the cells in the principal component space, providing a solid clustering foundation for subsequent cell type analysis B Detailed annotation of the cells in these 16 clusters. They can be categorized into eight distinct cell types: Inhibitory neurons, Excitatory neurons, Vascular leptomeningeal cells, Oligodendrocyte precursor cells, Oligodendrocytes, Perivascular macrophages (PVMs), Endothelial cells, and Astrocytes. These annotations provide an initial understanding of the biological significance behind each cluster. C The bubble plot illustrates the relationship between these eight different cell types and their corresponding cell markers. This plot provides an intuitive representation of the expression patterns of cell markers across each cell type, aiding in the confirmation of the biological characteristics and functions of these clusters. D The differences in the proportional representation of these eight cell types between the two sample groups
To identify Marker genes of PVMs, we used the FindAllMarkers function in Seurat with filtering criteria of ∣logFC∣ > 1|logFC|> 1∣logFC∣ > 1 and adj.Pval < 0.05adj.Pval < 0.05adj. Pval < 0.05, which were then used for subsequent research (cellMarkers.txt). The CellChat tool was applied to analyze the ligand-receptor interactions among the different cell subtypes, revealing complex communication networks (Fig. 2A). Specifically, PVMs exhibited close communication relationships with oligodendrocytes, astrocytes, oligodendrocyte precursor cells, and inhibitory neurons (Fig. 2B). Finally, pathway analysis of the PVM Marker genes was performed using the Metascape database, which identified significant involvement in processes such as actin filament-based processes, hemopoiesis, and GTPase regulator activity (Fig. 3).
Fig. 2
Cell communication A Cell interaction network diagram for the eight different cell types, where the edge width represents the probability and strength of communication between cells. This network diagram aids in understanding and visualizing the complex interactions between different cell types, revealing potential functional and regulatory networks among cells B Bubble plots to visualize receptor-ligand interactions between cells. These plots display the interaction patterns between different cell types, with the size of the bubbles representing the expression level or significance of the receptor-ligand pairs in each cell type. Through these plots, we can explore and analyze the molecular basis of cell–cell signaling and regulation, providing a visual and quantitative approach to understanding the complex interactions between cells
Fig. 3
Enrichment Analysis. We performed GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analyses using the Metascape database. These analyses helped identify significantly enriched biological processes, molecular functions, and cellular components, as well as key signaling and metabolic pathways within the gene sets involved in our study
MR Analysis o Identify Key Gene Predictors of ADMR analysis was used to establish key genetic predictors of AD by utilizing summary statistics from 488,285 samples, comprising 487,331 controls and 954 AD cases. The outcome data was retrieved with the ID ieu-b-5067. To select transcript genes for MR testing, we analyzed single-cell transcriptomic data to identify genes significantly associated with AD-associated PVM. Genes that reached the significance threshold of (p.adjust.value < 0.05, fold change > 1) were prioritized for MR testing. A total of 278 were evaluated, of which four gene pairs were screened as causally related to AD, based on the eQTL positive outcome, with statistical significance assessed using the Inverse Variance Weighted (IVW) method, where the p-value was set to < 0.05 (Fig. 4A–D). These gene pairs included IFNGR1, KLHL5, NUMB, and WDFY4. The causal effect estimates for each gene were as follows: IFNGR1 (0.9990; 95% CI: 0.9982–0.9997; p = 0.0071), KLHL5 (0.9996; 95% CI: 0.9990–0.9999; p = 0.0048), NUMB (0.9993; 95% CI: 0.9986–0.9999; p = 0.0365), and WDFY4 (0.9996; 95% CI: 0.9992–0.9999; p = 0.0277). These genes were identified as proactive factors in the development of AD, suggesting that they contribute significantly to the disease’s pathogenesis. To further evaluate the stability of these findings, a sensitivity analysis was conducted, which showed that the effect of removing any single SNP did not notably alter the causal estimates. This indicates that the relationships between the four gene pairs and AD are robust and not influenced by any single variant, thereby reinforcing the reliability of the results (Fig. 5A–D).
Fig. 4
Mendelian Randomization Analysis. A–D Scatter plots for the causal association between key genes and Alzheimer's disease(AD). In the plots, different colors represent various statistical methods, and the slope of the lines indicates the causal effect for each method
Fig. 5
Forest plots. Each forest plot represents a SNP corresponding to a key gene, displaying the results of the Leave-One-Out test. A The forest plot showed primary results of the causal association between IFNGR1 and Alzheimer's disease. B The forest plot showed primary results of the causal association between KLHL5 and Alzheimer's disease. C The forest plot showed primary results of the causal association between NUMB and Alzheimer's disease. D The forest plot showed primary results of the causal association between WDFY4 and Alzheimer's disease
Immune Infiltration AnalysisThis study further analyzed the association between key genes and immune infiltration to explore the molecular mechanism of AD. To account for the compositional nature of immune infiltration proportions, we applied Dirichlet statistical correction. The corrected proportions are illustrated in Fig. 6A, B, highlighting the relative abundance of immune cell subtypes across conditions. This adjustment ensures robust representation of the immune landscape. In addition, the results showed that the case group had a significantly higher level of Macrophages M2 than the control group. (Fig. 6C). This finding suggests that Macrophages M2 may be more prevalent in AD-affected tissues, potentially playing a critical role in the disease’s immune response and inflammatory processes.
Fig. 6
Immune Infiltration. A The relative percentages of 22 immune cell subsets in both the control group and the disease group. These data reflect the distribution differences of various immune cell subsets between the two groups, providing fundamental insights into the changes in the immune system under disease conditions. B Pearson correlation among the 22 immune cell subsets. Color coding represents the strength of different correlations: blue indicates negative correlations, while red represents positive correlations. This correlation analysis helps identify potential synergistic or competitive interactions between immune cell subsets. C The differences in immune cell content between the control group and disease group patients. Blue bars represent the average content in the control group, while pink bars represent the average content in the disease group. These bar charts clearly depict the changes in the quantities of different immune cell subsets between the two groups, revealing the impact of the disease state on the overall immune system and specific subsets. D Scatter plots display the correlation between each gene and the content of various immune cell subsets, helping to identify potential gene regulatory networks and their roles in immune modulation
Association Between Key Genes and Immune CellsThe association between key genes and immune cells was further investigated. Significant positive correlations were observed between IFNGR1 and T cells gamma delta, naïve B cells, Neutrophils, and regulatory T cells (Tregs). In contrast, a significant negative correlation was observed between IFNGR1 and memory B cells. In addition, significant positive correlations were observed between KLHL5 and resting NK cells, naïve B cells, and Tregs. Furthermore, NUMB showed significant positive correlations with memory B cells and naïve CD4 T cells, suggesting that these genes may influence immune responses by interacting with these immune cell populations. On the other hand, a significant negative correlation was observed between WDFY4 and CD8 T cells (Fig. 6D). Further analysis of the associations between key genes and various immune factors revealed significant correlations, indicating the crucial role of these key genes in the immune microenvironment. (Fig. 7A–E).
Fig. 7
Correlation between Key Genes and Immune Factors. A–E The correlation bubble plots illustrates the correlation between key genes (IFNGR1, KLHL5, NUMB and WDFY4) and chemokines (CXCL6, CXCL2, CCL3, etc.), immunoinhibitors (TGFBR1, TGFB1, CSF1R, etc.), immunostimulators (TNFRSF25, CD86, CD276, etc.), MHC molecules (HLA-DRA, HLA-DQA2, B2M etc.), and receptors (CCR10, CX3CR1, CXCR5, etc.). In these plots, each bubble represents the correlation between a key gene and the corresponding biological process
Construction of Transcriptional Regulatory Network and miRNA Network Related to Key GenesFurther analysis revealed that the key genes shared common regulatory mechanisms, involving various transcription factors (TFs). These TFs were enriched using cumulative recovery curves to identify the most relevant regulatory patterns. Motif-TF annotation and selection analysis highlighted cisbp__M6111 as the motif with the highest normalized enrichment score (NES) of 8.86, suggesting that this motif plays a significant role in the regulation of the key genes. Figure 8A, B display the complete list of motifs along with their corresponding TFs, providing insights into the potential regulatory networks at play. In addition, we utilized the miRcode database to retrieve the noncoding RNA network for the four key genes. This analysis predicted a total of 356 miRNAs and 394 associated gene-miRNA pairs, which were visualized through Cytoscape (Fig. 8C). This network further emphasizes the complex regulatory interactions between key genes and miRNAs, shedding light on the post-transcriptional regulation mechanisms involved in AD.
Fig. 8
Transcriptional Regulation and miRNA Network Associated with Key Genes A Transcriptional regulatory network diagram for key genes, where red represents the key genes and green represents transcription factors. Each red node indicates a key gene, while each green node represents a transcription factor potentially involved in regulating the key gene. This network diagram provides a visual representation of the regulatory relationships between key genes and transcription factors, helping to uncover the transcriptional regulatory mechanisms of the key genes. B The diagram illustrates the motifs enriched in key genes and their corresponding transcription factors. Each motif is associated with a specific transcription factor, and by examining the relationship between these motifs and transcription factors, we can gain insight into how these transcription factors may regulate the expression of key genes by binding to specific motifs. This information helps reveal the detailed mechanisms within the transcriptional regulatory network of key genes. C The miRNA network diagram of key genes, where circles represent mRNAs and inverted triangles represent miRNAs. Each circular node represents the mRNA of a key gene, while each inverted triangle node represents a miRNA that may regulate the expression of the mRNA by binding to it. This network diagram helps identify which miRNAs may influence the expression of key genes by binding to their mRNAs, providing insights into how miRNA-mediated regulation affects the function of key genes and contributes to understanding their roles in cellular regulation mechanisms
Expression Profile of Key Genes in Single-Cell Data and Associated Immune Metabolism Pathways/Disease GenesThis study analyzed the expression of key genes in single cells, focusing on IFNGR1, KLHL5, NUMB, and WDFY4. The results demonstrated the expression patterns of these genes across various cell types, including astrocytes, endothelial cells, PVM, oligodendrocytes, oligodendrocyte precursor cells, vascular leptomeningeal cells, excitatory neurons, and inhibitory neurons (Figs. 9A–C). To further investigate the involvement of these key genes in immune metabolism, we employed AUCell to quantify and score their expression within immune metabolism-related pathways in single cells. The gene expressions in these pathways were visualized using bubble charts, which highlighted the varying levels of expression for each key gene. Notably, the results indicated that WDFY4 was more active in cells with high expression of IL-6_JAK_STAT3 signaling in the immune pathway, as well as Notch signaling in the signaling pathway (Fig. 9D). These findings suggest that WDFY4 may play a significant role in modulating these critical immune and signaling pathways.
Fig. 9
Single-Cell Expression A–C present the expression profiles of key genes at the single-cell level. These charts provide insights into the expression levels and patterns of key genes across different cell types, aiding in a deeper understanding of the functions and regulatory networks of these genes at the single-cell level. D Correlation between Key Genes and Immune Metabolic Pathways. By analyzing the co-expression relationships between key genes and other genes in immune metabolic pathways, we can gain insights into the roles and regulatory mechanisms of these genes in immune metabolic processes. This understanding provides new perspectives and ideas for disease treatment and immune regulation
Subsequently, we investigated the specific signaling pathways associated with the key genes to explore the potential molecular mechanisms by which these genes influence disease progression. The GSEA results revealed that IFNGR1 is enriched in several signaling pathways, including the NF-κB, Toll-like receptor (TLRs), and TGF-β signaling pathway (Fig. 10A). KLHL5 is enriched in the TGF-β, Hippo, and Notch signaling pathway (Fig. 10B). NUMB is enriched in the Notch, NF-κB, and PI3K-Akt signaling pathway (Fig. 10C). WDFY4 is enriched in the Hippo, TGF-β, and Notch signaling pathway (Fig. 10D).
Fig. 10
GSEA analysis reveals enriched pathways in patient groups based on key gene expression. Gene Set Enrichment Analysis (GSEA) was applied to compare the enrichment of pathways between high and low expression groups of key genes. Higher Enrichment scores (ES) indicates stronger enrichment in the high-expression group. A GSEA analysis found that IFNGR1 expression was positively correlated with the NF-κB, Toll-like receptor (TLRs), and TGF-β signaling pathway B KLHL5 expression was positively correlated with the TGF-β, Hippo, and Notch signaling pathway C NUMB expression was positively correlated with the Notch, NF-κB, and PI3K-Akt signaling pathway D WDFY4 expression was positively correlated with the Hippo, TGF-β, and Notch signaling pathway
GSVA analysis indicated that IFNGR1 is enriched in signaling pathways such as INFLAMMATORY_RESPONSE and ALLOGRAFT_REJECTION (Fig. 11A). KLHL5 is enriched in the P53_PATHWAY and ALLOGRAFT_REJECTION pathways (Fig. 11B). NUMB is enriched in the MITOTIC_SPINDLE and APICAL_JUNCTION pathways (Fig. 11C). WDFY4 is enriched in the NOTCH_SIGNALING and ESTROGEN_RESPONSE_EARLY pathways (Fig. 11D). These findings suggest that the key genes may influence disease progression through these pathways.
Fig. 11
GSVA analysis of key signaling pathways in patient samples. Gene Set Variation Analysis (GSVA) was performed to assess the enrichment of key signaling pathways in different patient groups. Each bar represents the GSVA score for a specific gene set in a given sample, with the height indicating the level of enrichment. A IFNGR1 is enriched in signaling pathways such as INFLAMMATORY_RESPONSE and ALLOGRAFT_REJECTION. B KLHL5 is enriched in the P53_PATHWAY and ALLOGRAFT_REJECTION pathways C NUMB is enriched in the MITOTIC_SPINDLE and APICAL_JUNCTION pathways D WDFY4 is enriched in the NOTCH_SIGNALING and ESTROGEN_RESPONSE_EARLY pathways
Pseudo-Time Analysis and Developmental Trajectory of PVMWe calculated the similarity between PVM cells and constructed their cell differentiation trajectory. By visualizing this trajectory, we were able to generate a pseudo-time trajectory that illustrates the process of cell development. This trajectory image depicts cell differentiation along with the gene expression patterns at various time points, allowing us to trace the cellular progression over time. The trajectory was visualized with cells colored according to their pseudotime value (Pseudotime is a probability calculated by Monocle based on gene expression data, representing the temporal order of cell states), cell type, and state (with different cell states distinguished by path branches) (Fig. 12A, B). While genes exhibited differential expression across the trajectory’s branches, they could not be effectively displayed in an overall heatmap. To address this, we identified all the branch points, calculated genes with significant differential expression before and after these branch points, and visualized these changes in branch heatmaps (Fig. 12C). The trajectory begins in the center and differentiates outward, with the two branches representing the contrasting gene expression patterns at each end. The legend in the figure indicates that "Cell Type’s pre-branch" corresponds to the data collected before the branch point, "Cell fate1" refers to the side with a smaller state after the branch point, and "Cell fate2" represents the side with a larger state after the branch point. The heatmap was further divided into six distinct clusters based on gene expression changes across the trajectory. Additionally, the expression changes of the key genes, influenced by the cell status and other factors, are shown in Fig. 12D, offering insights into how these genes evolve throughout the cell differentiation process.
Fig. 12
Cell Developmental Trajectory A, B present the pseudotime analysis and developmental trajectory of cells. These charts reveal the changes in cell states and developmental pathways over time by analyzing gene expression patterns in single cells throughout the developmental process. This analysis helps us understand the dynamic progression of cell development and the regulatory roles of key genes at different developmental stages. C reveals the gene expression dynamics of each PVM cell branch. By analyzing the expression patterns and dynamic changes of genes in different PVM cell branches, we can gain insights into the developmental processes and functional characteristics of these branches. This provides important clues for studying the development and function of PVM. D The relationship between key genes expression and PVM developmental trajectory. By analyzing the expression patterns and changes of key genes during the PVM development process, we can uncover the roles and regulatory mechanisms of these genes in PVM development. This provides important insights and references for understanding the molecular mechanisms underlying PVM development
Comments (0)