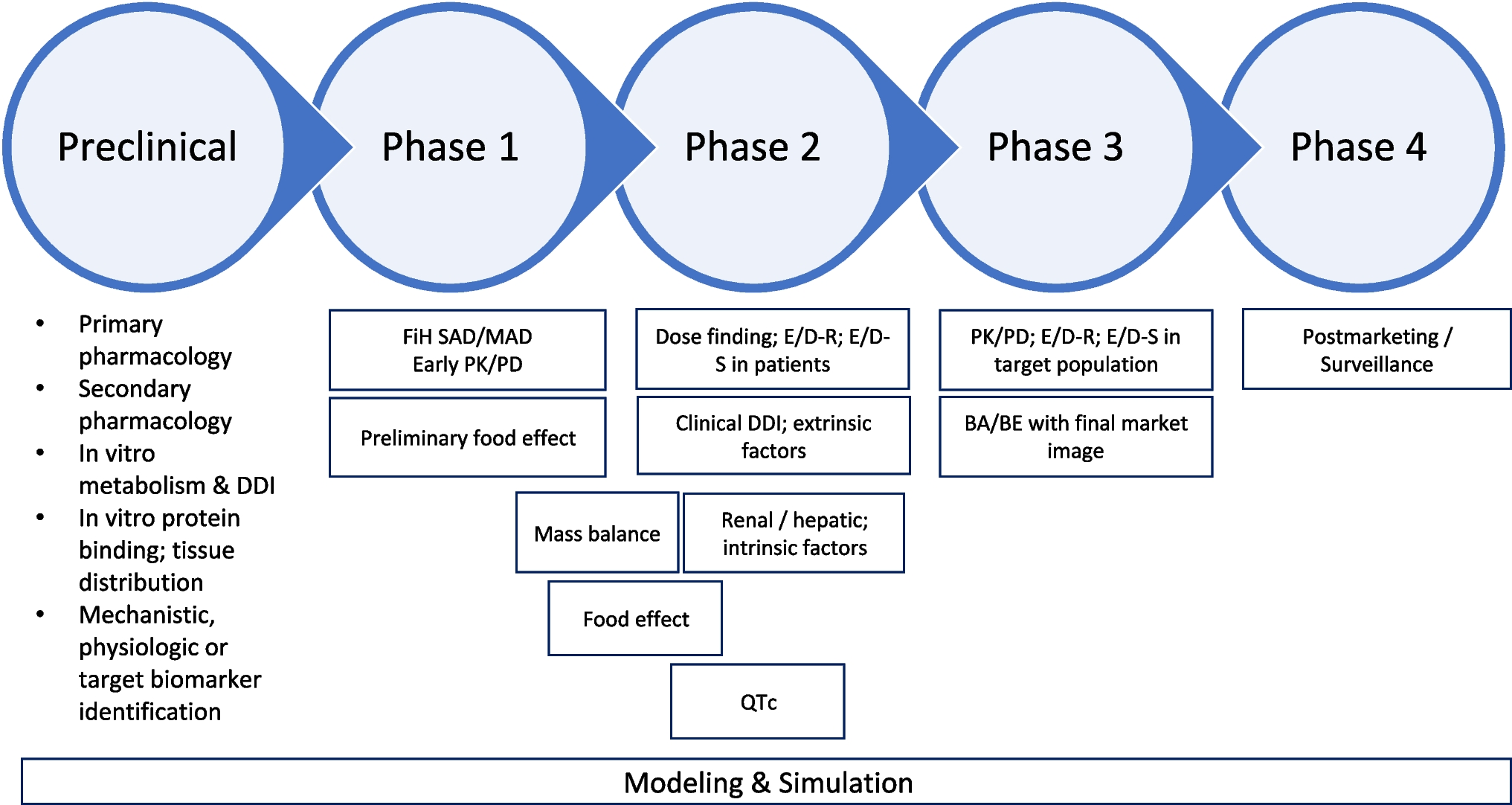
Over the past decade, we as consultants have recognised a trend towards a higher integration and more complex study designs in the early clinical trials. Not only that single and multiple ascending dose (SAD/MAD) studies are more often combined in one study protocol but also are enriched with other study types such as elderly and/or ethnic subgroup cohorts, drug-drug interaction (DDI), or food effect assessments. If well planned and accompanied by clear decision rules, this can expedite the development process and increase the efficiency of drug development. Another trend is that developers want to test their assets in the patient earlier than at times when the development path was more step-wise. Early clinical readouts are important for research teams to make decisions and for investors to fund further research. However, this approach bears the risk that the effect size, the variability in the data, and the sample size in early clinical trials do not match well. An initial ‘mini’ proof-of-concept study does not replace a properly designed Phase II study. On the other hand, pharmacodynamic effects related to biomarkers may be overinterpreted and require a solid understanding of the role of the biomarker in the research project including its proper incorporation into the early clinical trial [10, 11].
Dose optimisation too often is an afterthought because the full spectrum of data is not always exploited at the time it becomes available. We sometimes get asked how we would justify an ‘optimal’ dose at a later stage of development. The general fear of missing the therapeutic effect may result in using an inappropriately high dose that works well in the indication. Often, ‘less is more’ may be the better option (i.e. going with a lower dose) by fully analysing safety, pharmacodynamics, and efficacy data in an integrated way. MIDD-based approaches help integrate data from multiple sources in the form of computational models based on the understanding of physiology, pharmacology, and disease processes. Applying these models informs drug development decision-making and registration interactions, especially with respect to optimisation of the design of future clinical studies, dose regimen optimisation, and individualisation [9]. Under the sixth iteration of the Prescription Drug User Fee Act (PDUFA VI), FDA committed to developing MIDD-related guidance updates, holding public workshops on these approaches, and establishing a standard operating procedure for reviews of MIDD-related submissions. In 2018, FDA also established the MIDD Pilot Program [12], which affords sponsors or applicants the opportunity to meet with agency staff to discuss modelling approaches in medical product development. In its regulatory science strategy (mid-point achievement report) the European Medicines Agency (EMA) desires to enhance modelling and simulation and extrapolation use across the product lifecycle and leverage the outcome of EU projects and to optimise their own capabilities in modelling, simulation, and extrapolation [13].
Certainly, utilising dose response information to support dose selection and optimisation is not a new concept. The ICH Harmonised Guideline E4 was finalised under Step 4 in March 1994 and adopted worldwide by all major regulatory bodies [14]. This document gives recommendations on the design and conduct of studies to assess the relationships among dose, drug-concentration in blood, and clinical response throughout the clinical development of a new drug. However, since then, evolutions in study designs (e.g. adaptive/seamless designs, model informed adaptations) and methods for characterising the dose/exposure–response relationship (e.g. pharmacometrics, quantitative systems approaches, Bayesian approaches) led to a new era of methodological applications and practices used in drug development [15].
Nowadays, we have many more tools in our hands than 30 years ago, but has it become easier to get the necessary analyses done? The biopharmaceutical landscape and industry interests have changed, and developers, regulators, and we as clinical pharmacologists deal more often with confirmatory trials in small populations (paediatrics, rare/orphan diseases) as well as expedited development programmes, with a ‘backloaded’ rather than ‘frontloaded’ approach, leading to a reduction in data pre-approval and a shift of data collection to the post-approval stage. With limited numbers of patients, testing multiple dose levels and regimens remains challenging and makes an informed dose decision one of the major challenges in rare disease drug development. Clinical pharmacology plays a strategic role in bridging this gap, providing valuable insights that support both the drug development process and regulatory approvals [16,17,18]. In an evaluation of new drug applications (NDA) in approved drugs for the treatment of rare diseases, modelling and simulation approaches were utilised to address many clinical pharmacology questions, with population pharmacokinetic analyses used extensively in the evaluation of the effect of organ impairment on drug exposure and with physiologically based pharmacokinetic analyses used mainly in assessing drug interaction risks [19].
Dose individualisation has become crucial in modern healthcare, particularly for therapies in oncology [20, 21]. It aims to maximise treatment benefits while reducing side effects. Traditionally, doses were personalised in drugs with a narrow therapeutic index by therapeutic drug monitoring (TDM), best known for digoxin or theophylline. TDM has been increasingly used in the past years to guide dose adjustments mostly in oncology [22]. The one-size-fits-all fixed dosing approach is still the standard for most drugs but may be questioned the more information we have on pharmacogenomics and patient characteristics and understand about genetic differences. Tools such as Bayesian or population pharmacokinetic models and TDM are helpful in making dosing decisions, particularly if they are combined [22, 23].
Despite challenges and a need for flexibility and adoption, we as clinical pharmacologists believe that there are some fundamental principles as a mainstay of drug development. The exposure–response relationship assessment is a key effort that always needs to be undertaken.


Comments (0)