
Remember me
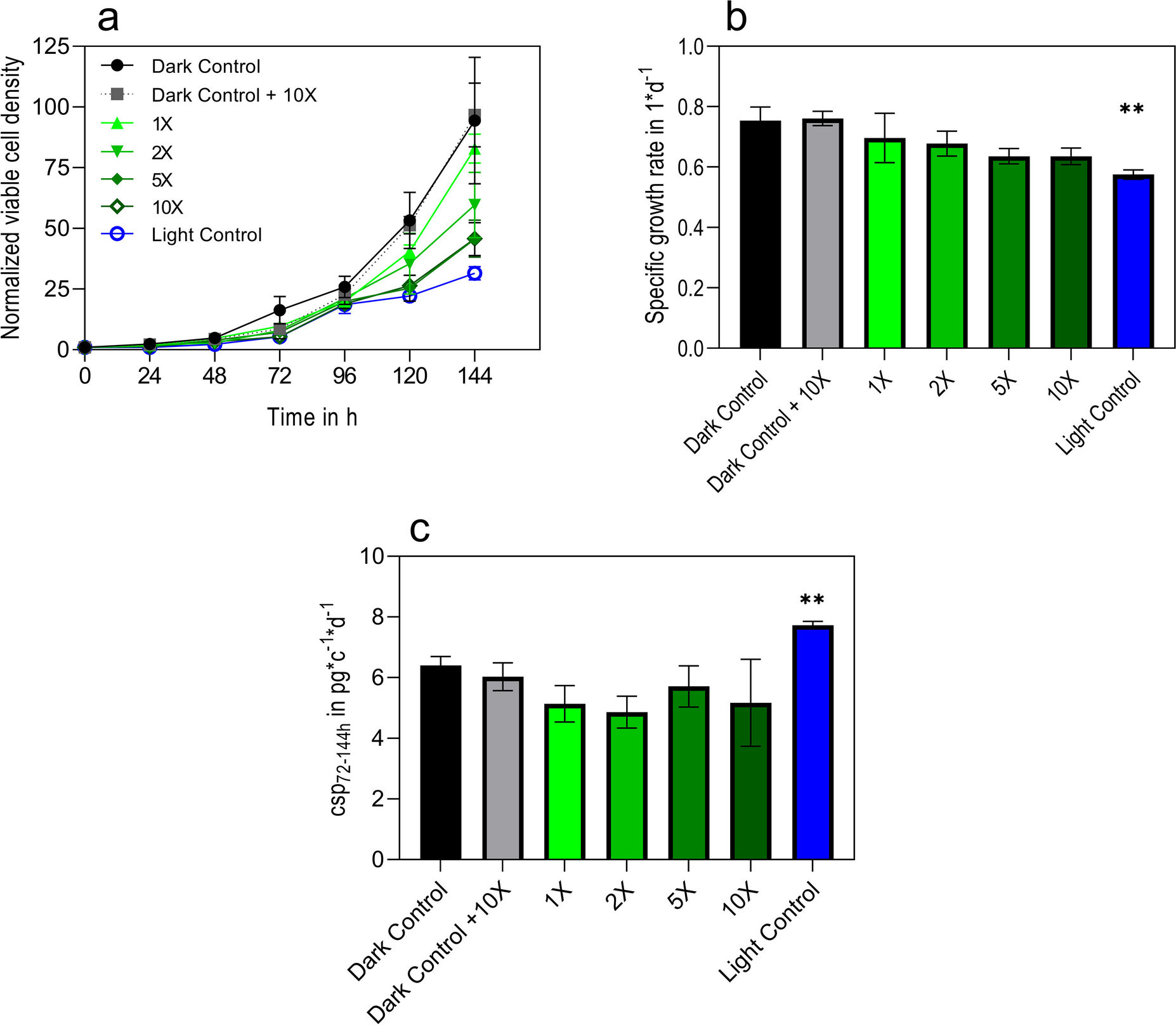

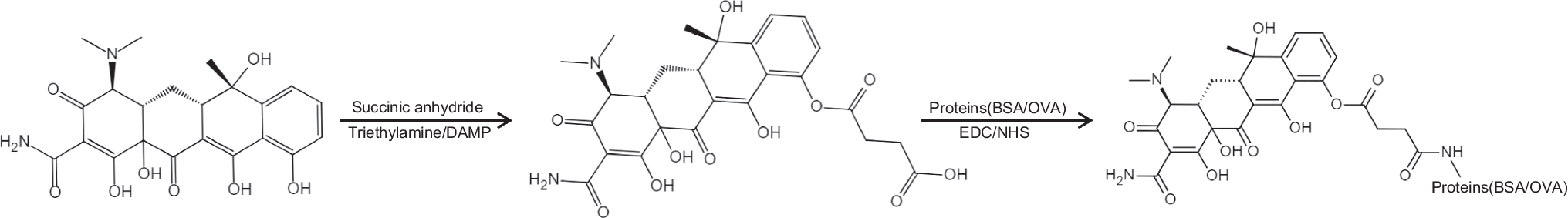
A cross-sectional cohort study was conducted between June and August of 2023 in the Brazilian Amazon. The study enrolled 323 participants residing in malaria-affected municipalities of Cruzeiro do Sul (141), Mâncio Lima (72), and Guajará (61) (Fig. 2). Cruzeiro do Sul, situated at 07° 37′ 50″ S/72° 40′ 13″ W, and Mâncio Lima, located at 07° 36′ 49″ S/72° 53′ 47″ W, are both recognized as high-risk areas within the Juruá Valley of Acre State, renowned as Brazil’s primary hotspot for P. falciparum malaria. Guajará, positioned at 02° 58′ 18″ S/57° 40′ 38″ W, is designated as a medium-risk area within Amazonas State (Malaria-Brasil 2023).
Fig. 2
Brazil map showing studied areas in the spotlight. Areas of malaria transmission in Brazil and studied areas according to the Annual Parasitary Index (API, number of autochthonous cases per 1000 inhabitants. Very low API indicates that there are less than 1 case/1000 inhabitants, low API indicates that there are less than 10 cases/1000 inhabitants, medium API indicates 10–49.9 cases/1000 inhabitants and high API more than 50 cases/1000 inhabitants (Malaria-Brasil 2023)
Epidemiological surveyIndividuals who seek medical attention in health care facilities for malaria screening and treatment, and from their contacts and neighbors were enrolled in the study. Written informed consent was obtained from all adult donors who consented to participate or from the donor’s parents in the cases of children. Donors who consented to participate also completed an epidemiological survey aimed to assess the extent of malaria exposure. Participants answered questions regarding personal information such as age, time of residence in a malaria-endemic area, history of previous malaria episodes, time since the last infection, use of malaria prophylaxis, and presence of symptoms.
Blood sampling and malaria diagnosisAfter obtaining consent and completing an epidemiological survey, 10 mL of venous peripheral blood samples were collected in ethylenediamine tetraacetic acid (EDTA) tubes (Becton Dickinson, Franklin Lakes, New Jersey, USA). These samples underwent immediate mixing with an equal volume of cryopreservation solution containing 0.9% NaCl, 4.2% sorbitol, and 20% glycerol, ensuring optimal preservation. Subsequently, the samples were stored at − 70 °C until required for further analysis.
During the clinical interview, patient samples were submitted to the Bioline Malaria Ag Pf/Pf/Pv Rapid Diagnostic Test (Abbott, Chicago, Illinois, USA). Results were annotated after 15 min, according to the manufacturer’s instructions. Malaria was also diagnosed by examination of 200 fields at 1.000 × magnification under oil immersion in Giemsa-stained thick and thin blood smears. Thin blood smears of the positive samples were examined for species identification by a skilled technician with extensive experience in malaria diagnosis at the Laboratory of Malaria Research (FIOCRUZ, Rio de Janeiro, Rio de Janeiro, Brazil), which serves as the headquarters of the CEMART (Centre for Malaria Research and Training), recognized as a reference center for malaria diagnosis in the extra-Amazonian for the Brazilian Ministry of Health. Positive individuals for P. vivax and/or P. falciparum at the time of blood collection were subsequently treated with the chemotherapeutic regimen recommended by the Brazilian Ministry of Health (Brasil 2020).
DNA extractionFor the molecular diagnosis of malaria, DNA was isolated and purified from 200 µL blood samples by using the QIAamp™ DNA Blood Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. The extracted DNA was promptly stored at − 20 °C until subjected to nested PCR (nPCR) or qPCR.
Nested polymerase chain reaction (nested PCR)PCR using specific primers for the Plasmodium genus (Plasmodium spp.) and the species P. falciparum and P. vivax was performed according to the method described by Snounou et al. (1993). The first PCR detects the Plasmodium genus, while the second one differentiates between the Plasmodium species using 2 pairs of internal primers (GenOne Biotechnologies, Rio de Janeiro, Rio de Janeiro, Brazil) specific to the Plasmodium species targeted under the study (Table 1). For the first PCR, 2 μL of DNA was amplified in a reaction mixture containing 1X buffer, 0.8 μM of each primer, 0.2 mM dNTPs (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 1.5 mM MgCl2, and 1 unit of AmpliTaq Gold™ DNA polymerase (Thermo Fisher Scientific, Waltham, Massachusetts, USA). For the second PCR, 2 μL of amplified DNA from the first PCR was added to the reaction mixture containing the species-specific primers (Table 1). The nested PCR reactions were conducted using a ProFlex™ PCR System (Applied Biosystems, Waltham, Massachusetts, USA). Each reaction batch included negative controls (DNA extracted from non-infected blood), no DNA template controls (PCR-grade ultrapure water), and positive controls (DNA extracted from P. falciparum culture or P. vivax isolates). All PCR products were analyzed by 2% agarose gel electrophoresis in 1X TAE buffer (Tris–acetate 0.04 M, EDTA 1 mM) with 0.5 μg/mL ethidium bromide (Sigma-Aldrich, St. Louis, Missouri, USA). PCR products were visualized under ultraviolet (UV) light, and product sizing was performed using GeneRuler™ 100 bp and 1 Kb DNA Ladder (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Table 1 Primers, probes, and PCR cycling steps used to perform the detection of Plasmodium speciesQuantitative real-time PCRSamples were also submitted to qPCR for detection of Plasmodium spp., P. falciparum, and P. vivax using primer and probe sets (GenOne Biotechnologies, Rio de Janeiro, Rio de Janeiro, Brazil) previously described by Hassanpour et al. (2016), Perandin et al. (2004), and Almeida-de-Oliveira et al. (2019) for the genus, P. falciparum, and P. vivax, respectively. Negative controls (DNA extracted from non-infected blood), no DNA template controls (PCR-grade ultrapure water), and positive controls (DNA extracted from P. falciparum culture or P. vivax isolates) were used in all reaction plates. The primers, probes, and qPCR cycling steps used are listed in Table 1.
All qPCR experiments were performed with a final reaction volume of 20 µL, consisting of 2 µL template DNA, 1 μL of each primer (900 nM), 1 μL probe (250 nM), 6 µL of 1X TaqMan™ Universal Master Mix II with UNG (Applied Biosystems, Waltham, Massachusetts, USA), and 10 μL UltraPure™ distilled water (Applied Biosystems, Waltham, Massachusetts, USA). Amplifications were conducted in MicroAmp™ optical 96-well reaction plates (Applied Biosystems, Waltham, Massachusetts, USA) using a QuantStudio™ 3 Real-Time PCR System (Applied Biosystems, Waltham, Massachusetts, USA), and the results were analyzed with QuantStudio™ Design and Analysis Software v1.5.2 (Applied Biosystems, Waltham, Massachusetts, USA). Samples with a qPCR Cq value greater than 37.5 cycles were considered inconclusive and were excluded from the analysis.
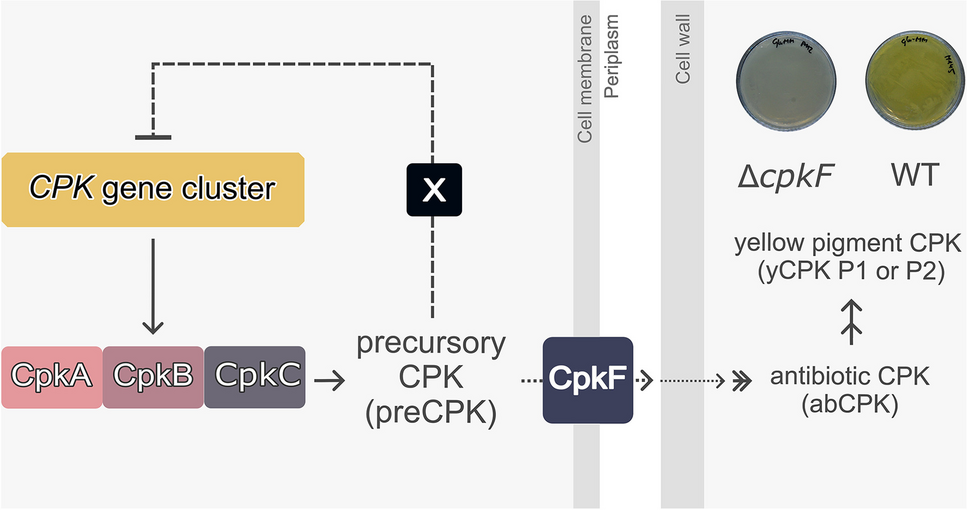
GENEYE® ERA Plasmodium detection kit assayThe GENEYE® ERA Plasmodium detection kit assays were performed directly from whole blood, without requiring a DNA purification step. The primers and FAM/BHQ1 probes (GenOne Biotechnologies, Rio de Janeiro, Rio de Janeiro, Brazil) were designed to target the 18S rDNA, based on a consensus of reference genome sequences for Plasmodium spp. (NC_004331.3: 2800005–2802154, NC_037282.1: 1925995–1928144, NC_009915.1: 13208–11933, NC_009908.2: 806754–808029), with specific sequences for P. falciparum (NC_004331.3: 2800005–2802154) and P. vivax (NC_009908.2: 806754–808029). The workflow includes pre-warming the device, setting up the apparatus and software, configuring detection settings, sample preparation, adding the sample lysate to the test tube, conducting the incubation and detection phase, and monitoring the fluorescence readings (Fig. 3). The following protocol provides comprehensive details on each executed step.
Fig. 3
Schematic workflow of GENEYE® ERA Plasmodium detection kit. WB: whole blood, A: Buffer A, M: Buffer M, L: sample lysate. 1. Pre-warm the device by heating the bath to 40 °C; 2. Turn on and connect the isothermal device to the app; 3. Organize and prepare all necessary reagents; 4. Add 20 µL of whole blood to 2 mL of buffer A; 5. Carefully homogenize the tube and let it incubate at room temperature for 5 min; 6. Add 40 µL of lysate to the bottom of the piston tube, followed by 10 µL of buffer M to the piston tube’s inner bottom wall; 7. After closing the piston tube cap tightly, add 50 µL of buffer M to the top cavity of the piston tube and mix it carefully by hand; 8. Place the piston tube in the dry bath at 40 °C for a 15-min incubation period; 9. Press the pistons to mix the buffer M and the bottom pre-mix, then remove the top of the piston tube; 10. Insert the piston tube into the GENEYE® Mini Isothermal ERA device; 11. After 5 min, check the fluorescence readings on the app to determine if the result is “negative” or “positive”
Pre-warmingA water bath or dry bath (heat block) was pre-warmed to a temperature of 40 °C. The GENEYE® Mini Isothermal ERA device was then turned on, and the pre-warming phase was allowed to complete, as indicated by the “Ready” status on the display panel.
Device setupThe “GENEYE” mobile application (app) (GenDx Biotech Suzhou, China) was released as a software application on smartphones. The user account was logged into the device, and access to the detection section was obtained. Subsequently, testing was initiated within the “Malaria testing” section. The Bluetooth on the phone was enabled, and the “GENEYE” device was selected for connection. Upon selecting the device name, the connection was established.
Setting configurationThe configuration setting parameters for malaria detection were adjusted. Specifically, the Fluorescence Difference Multiple was set to “10,” the Reaction Temperature was set to “40 °C,” and the Detection Points were specified as “180.”
Sample preparationA volume of 20 µL of cryopreserved whole blood was added to a microtube containing 2 mL of buffer A. After closing the cap tightly, the tube was shaken thoroughly and left at room temperature for 5 min to allow the complete release of the genetic material.
Sample lysate addition to piston tubeA volume of 40 µL of buffer A sample lysate was added to the bottom of the piston tube, followed by the addition of 10 µL of buffer M to the piston tube’s wall. After the piston tube cap was tightly closed, 50 µL of buffer M was added to the piston tube’s top cavity and properly closed with the testing piston. The piston tube was then manually homogenized for a brief period in order to ensure thorough mixing.
Reaction step 1 (incubation)The piston tube was promptly placed into the preheated dry bath at 40 °C and allowed to stand for 15 min to permit enzyme activation and initial target amplification.
Reaction step 2 (detection)The “start detection” button within the app was pressed to initiate the testing process. After the 15-min incubation period, the piston tube was pressed inwards to allow the 50 µL of buffer M and revealing reagent to reach the bottom of the piston tube and mix thoroughly, thereby initiating an amplification boost. Subsequently, the top of the piston tube was removed, and the piston tube was promptly placed into the GENEYE® Mini Isothermal ERA device. Following a 5-min incubation period, the app displayed a real-time fluorescence curve of the test, thereby enabling the determination of the results as either “negative” or “positive” based on the endpoint fluorescence value. A fluorescence value of ≥ 1000 was considered a positive result, whereas a value of < 1000 was considered a negative result.
During the GENEYE® ERA Plasmodium detection kit assay reactions, in addition to the results displayed on the app, the fluorescence readings were captured every minute at ten predetermined time points. These readings were analyzed to redundantly monitor the progression of the amplification process in real time. Samples that reached the threshold of 1000 after 5 min or displayed an increasing positive profile in the readings over time, although they did not reach 1000 by the T10 point (10 min), were considered inconclusive.
Statistical analysisThe data was entered into Microsoft Excel (Microsoft Corporation, Redmond, Washington, USA) and subsequently transferred to MedCalc (MedCalc Software Ltd, Ostend, Belgium) for detailed statistical analysis. Probit modeling was used to estimate the limit of detection (LoD) for the GENEYE® ERA Plasmodium detection kit, based on the detection data across various parasite densities, determining the density range at which 95% of samples was detected. The efficacy of each diagnostic tool, comprising the nPCR, the GENEYE® ERA Plasmodium detection kit, the qPCR, the microscopy, and the RDT, was evaluated though assessment of sensitivity (effectiveness at detecting the presence of Plasmodium spp., P. falciparum, and P. vivax, when it is present, with higher values indicating better detection capabilities); specificity (ability to rule out the presence of Plasmodium when absent, with higher values reflecting better exclusion of non-infected individuals); positive predictive value (PPV, likelihood of true positive results, with higher PPV indicating that positive test results are true); negative predictive value (NPV, reliability of negative results, with higher NPV suggesting that negative results are reliable); accuracy (overall correctness of the test); positive likelihood ratio (PLR, probability of positive results in infected individuals relative to non-infected individuals, with a PLR greater than 1 suggesting the test result is more likely in infected individuals); and negative likelihood ratio (NLR, probability of negative results in infected vs. non-infected, with an NLR less than 1 supporting the absence of Plasmodium when the test is negative). Additionally, the Cohen’s kappa statistic is employed to assess the degree of agreement between the observed results and the expected outcomes, beyond what would be expected by chance, and is therefore a useful metric to consider when evaluating test accuracy. The highly sensitive nPCR was employed as the reference standard, with the values obtained from the calculation of Cohen’s kappa statistic interpreted according to the following scale: ≤ 0 indicates no agreement, 0.01–0.20 slight agreement, 0.21–0.40 fair agreement, 0.41–0.60 moderate agreement, 0.61–0.80 substantial agreement, and 0.81–1.00 almost perfect agreement, to perfect agreement.
Comments (0)