Strains and media
The X. dendrorhous type strain CBS 6938 (ATCC 96594) was cultured at 21 °C in YPD (YEP, Sunrise Science, Knoxville, TN, USA, 1877-1 KG) plus 20 g/L glucose, Alfa Aesar, Haverhill, MA, USA, A16828) media for all experiments. For all light plate apparatus experiments, the X. dendrorhous seed cultures were shielded from any external light by wrapping the flasks with aluminum foil. X. dendrorhous transformants were selected using YPD with 30 mg/L nourseothricin (Jena Bioscience, Jena, Germany, AB-101-10ML). Solid media plates were made using 20 g/L agar (Sunrise Science, Knoxville, TN, USA, 1910-1 KG). Golden gate (Type IIS) cloning was conducted using chemically competent Escherichia coli DH5α cells (NEB, Ipswich, MA, USA, C2987H). Construction of destination vectors with the toxic selection marker ccdB was done with chemically competent One Shot ccdB Survival 2 T1R E. coli cells (Invitrogen, Waltham, MA, USA, A10460). Both E. coli strains were grown in 25 g/L LB Miller broth (Fisher Scientific, Hampton, NH, USA, BP1426-2) at 37 °C. Antibiotic selection was carried out using 100 mg/L carbenicillin (Alfa Aesar, Haverhill, MA, USA, J61949), 25 mg/L chloramphenicol (Alfa Aesar, Haverhill, MA, USA, B20841), and 50 mg/L kanamycin (Alfa Aesar, Haverhill, MA, USA, J61272) for level 0, level 1, and level 2 assemblies, respectively.
gDNA isolation and sequencing
Isolation of gDNA from X. dendrorhous and next-generation sequencing was performed as described by Collins et al. (2021). Benchmarking universal single-copy ortholog (BUSCO) analyses were conducted on nine publicly available X. dendrorhous genomes using basidiomycota_odb10 for a lineage, Augustus 3.2 for gene prediction, and Ustilago maydis as the Augustus species (Stanke et al. 2006; Manni et al. 2021a, b; Manni et al. 2021a, b). Genomes were sourced from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) and assembly identifiers were as follows; GCA_001007165.2 (Sharma et al. 2015), GCA_001600435.1 (RIKEN Center for Life Science Technologies, Wako, Japan 2016), GCA_001579715.1 (Bellora et al. 2016), GCA_014706385.1 (Gómez et al. 2020), GCA_019201825.1 (Xiamen Canco Biotech Co., Fujian, China 2021), GCA_023566135.1 (Nanjing Tech University, Nanjing, China 2022), GCA_028533015.1 (Luna-Flores et al. 2022), GCA_022059005.1 (Jilin Agricultural University 2022), and GCA_037127225.1 (this study).
Construction of a light plate apparatus for various light wavelengths
The light plate apparatus (LPA) was assembled following a user’s manual published by the Tabor lab at Rice University (Gerhardt et al. 2016). The LPA is an instrument capable of shining two individual LED lights on cell cultures in a 24-well plate. It is comprised of a 3D-printed shell surrounding a soldering board with 48 LED light sockets oriented below a 24-well plate with a clear bottom (Arctic White LLC, Lehigh Valley, PA, USA, AWLS-324042). The LPA allows each culture to be exposed to two unique LED lights without disrupting neighboring cultures.
RNA isolation from the light plate apparatus
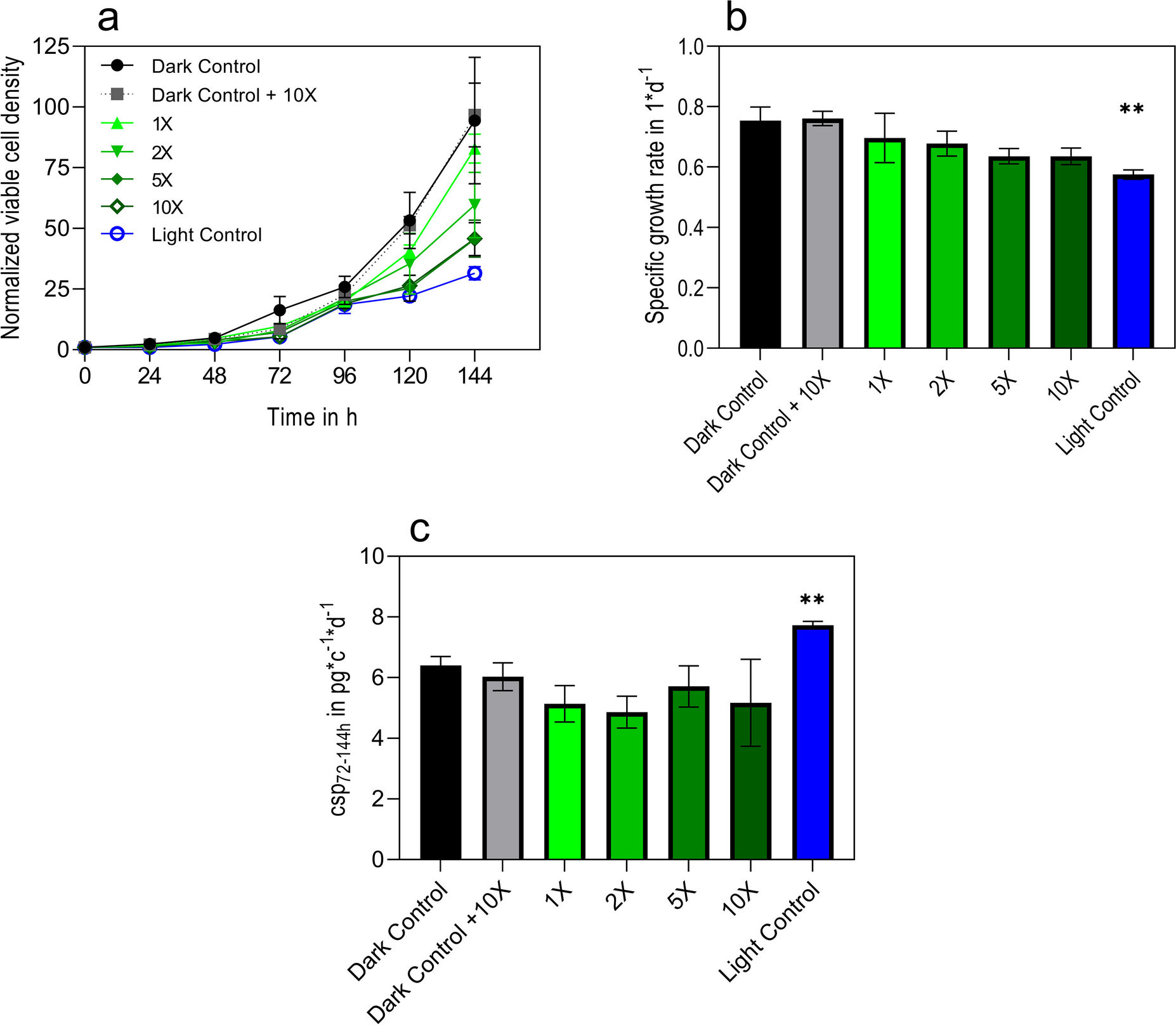
LPA cultures were started in 2 mL of YPD media at OD600 = 1 and shaken for 4 h at 200 rpm while exposed to one red (λ = 660 nm), green (λ = 565 nm), blue (λ = 470 nm), white (visible light spectrum), or UV LED light (λ = 400 nm), or no LED light. For the transcriptome sequencing experiments, the hydrogen peroxide condition replaced the white light condition. The hydrogen peroxide condition consisted of YPD media with hydrogen peroxide at a concentration of 10 mM and had no LED light. Then, 1 mL of X. dendrorhous culture was taken forward for RNA isolation, which used a cell homogenization and TRIzol-based method. X. dendrorhous cells were first resuspended in 200 μL of cell lysis buffer (0.5 M sodium acetate, 5% sodium dodecyl sulfate (SDS), and 1 mM ethylenediaminetetraacetic acid (EDTA)) and transferred to pre-filled tubes with 400-micron zirconium beads (OPS diagnostics, Lebanon, NJ, USA, PFMB 400–100-34). The cells were homogenized using a FastPrep-24 machine from MP Biomedical, Irvine, CA, USA, for two cycles at 6 m/s for 30 s each, with a 1-min incubation on ice between each cycle. Then, 800 μL of TRIzol reagent (Invitrogen, Waltham, MA, USA, 15,596,026) was then added to the cell lysate and incubated on ice for 10 min, followed by two more cycles on the FastPrep machine. On ice, 100 μL of 1–bromo–3–chloropropane (Sigma Aldrich, Burlington, MA, USA, B9673-200ML) was added to the cell lysate, inverted to mix, and incubated for 5 min. The tubes were spun down at 4 °C for 14,000 × g for 15 min. Then, 400 μL of supernatant was transferred to 400 μL of 100% ethanol. The RNA was purified using the Monarch Total RNA Miniprep Kit (NEB, Ipswich, MA, USA, T2010S) following the supplier’s instructions.
qRT-PCR evaluation of carotenogenesis pathway activity in response to light exposure
RNA samples were converted to cDNA using the Protoscript II First-Strand cDNA Synthesis Kit (NEB, Ipswich, MA, USA, E6560S) following the supplier’s standard protocol instructions. RT-qPCR was then performed using IDT’s PrimeTime Gene Expression Master Mix (Integrated DNA Technologies Inc., Coralville, IA, USA, 1,055,772) and PrimeTime qPCR pre-mixed assay with probes and primers. Sequences can be found in Supplemental Table S1. Probes targeting genes-of-interest were labeled with fluorescein amidite (FAM) fluorophores and probes targeting the actin housekeeping gene were labeled with hexachlorofluorescein (HEX) fluorophores. qRT-PCR was performed in triplicate. Primers were designed on Benchling (https://benchling.com/) and optimal probe placement was determined using PrimerQuest (https://www.idtdna.com/PrimerQuest). The QuantStudio 6 Flex system (Applied Biosystems, Waltham, MA, USA, 4,485,699) was used, and instructions provided by Integrated DNA Technologies for the standard cycling protocol were followed. qRT-PCR bar graphs were generated using GraphPad Software, Boston, MA, USA (www.graphpad.com).
RNA transcriptome sequencing and differential gene expression analysis
cDNA preparation and sequencing were performed per JGI’s standard workflow. Raw FASTQ file reads were filtered and trimmed using the Joint Genome Institute (JGI) QC pipeline (Clum et al. 2021). Filtered reads from each library were aligned to the reference genome using HISAT version 0.1.4-beta (Kim et al. 2015). featureCounts (Liao et al. 2014) was used to generate the raw gene counts file using gff3 annotation. Raw gene counts were used to evaluate the level of correlation between biological replicates using Pearson’s correlation (Benesty et al. 2009) and determine which replicates would be used in the differential gene expression (DGE) analysis. DESeq2 (version 1.10.0) was subsequently used to determine which genes were differentially expressed between pairs of conditions (Love et al. 2014). The parameter used to call a gene differentially expressed between conditions was a p-value < 0.05. Three biological replicates were performed for each treatment. Promoter names were determined by comparing gene annotations across ERGO’s gene ontology, Interpro, JGI predictions, and BLAST results, as shown in Supplemental Table S2. Heatmaps were generated through GraphPad Software, Boston, MA, USA (www.graphpad.com) and Adobe Illustrator (https://adobe.com/products/illustrator). Microsoft Powerpoint (https://office.microsoft.com/powerpoint) was used to generate protocol figures.
Gene set enrichment analysis
Annotated X. dendrorhous CBS 6938 genome and gene count table from mapped transcriptomic reads were uploaded to ERGO 2.04 (Igenbio, Chicago, IL, USA) (Overbeek 2003). Genes were functionally enriched according to ERGO groups using GAGE5 (Luo et al. 2009).
Polymerase chain reaction (PCR)
All PCRs were done using Q5 2X Master Mix (NEB, Ipswich, MA, USA, M0492L). Primers were designed on Benchling (https://benchling.com/). The OligoAnalyzer tool from Integrated DNA Technologies (IDT) (https://www.idtdna.com/pages/tools/oligoanalyzer) and the NEB Tm Calculator (https://tmcalculator.neb.com/) were used to check primer features. All primers were ordered from Integrated DNA Technologies, Coralville, IA, USA. PCR reactions closely followed NEB instructions. Briefly, reactions were done in 50 μL total volume; 25 μL Q5 Master Mix, 2.5 μL of each primer, X μL of template DNA (1 ng plasmid DNA or 100 ng genomic DNA), and 20-X μL nuclease free water (VWR, Radnor, PA, USA, 02–0201-0500). Reactions were run on a thermocycler following the manufacturer instructions.
Gibson cloning
Gibson assembly was used to construct destination vectors, which were designed for Type IIS-based cloning. Benchling (https://benchling.com/) was used to simulate Gibson assemblies. Reactions closely followed instructions from the NEBuilder HiFi DNA Assembly Master Mix (NEB, Ipswich, MA, USA, E2621S). Fragments were amplified with PCR and designed to have 20–30-bp overlaps. The DpnI enzyme was used to digest template plasmid or X. dendrorhous genomic DNA according to the manufacturer’s instructions (NEB, Ipswich, MA, USA, R0176S). Calculations and conversions were performed with aid of NEBioCalculator (https://nebiocalculator.neb.com). The amplified fragments were purified using the Zymo Clean and Concentrator Kit (Zymo, Irvine, CA, USA, D4005), followed by dilution to 0.2 pmols for 2–3 fragments or 0.5 pmols for 4 or more fragments. The fragments, 10 μL of HiFi master mix, and nuclease free water were combined in a PCR tube for a 20-μL total reaction volume. The mixture was run on a thermocycler at 50 °C for 60 min, with a hold at 10 °C.
Type IIS cloning and parts
A modular, hierarchical Type IIS cloning scheme was used to construct genetic designs for integration into X. dendrorhous (Iverson et al. 2016). This scheme consisted of three levels: transcriptional parts (level 0), transcription units (level 1), and integrative pathways (level 2). Cloning reactions were based on the enzymes BbsI (Thermo Scientific, Waltham, MA, USA, ER1011) or BsaI (NEB, Ipswich, MA, USA, R3733). Benchling (https://benchling.com/) was used to simulate Type IIS reactions. Cloning reactions consisted of 1 μL of N parts, 1 μL of BsaI or BbsI, 1 μ L of 10X Ligase Buffer, 0.4 μL of T4 DNA ligase (Promega, Madison, WI, USA, M1794), and 7.9 − N μL of nuclease free water, where N is the number of parts. Reactions were run on a thermocycler at 37 °C for 5 h, 50 °C for 15 min, 80 °C for 20 min, and a hold at 10 °C. All genetic parts used in this study can be found in Supplemental Table S3. All cloning vector maps can be found in Supplemental Fig. S1.
Minimum inhibitory concentration assay
X. dendrorhous cultures were diluted with YPD to an OD600 = 1.0 and 100 μL was pipetted into columns 2–11 of a 96-well plate (Corning, Corning, NY, USA, 3596). Column 11 served as a positive control of only X. dendrorhous culture. A negative control of 100 μL YPD was pipetted into column 12. The antifungals hygromycin (ThermoFisher, Waltham, MA, USA, 10,687,010), zeocin (Jena Bioscience, Jena, Germany, AB-103S), geneticin (ThermoFisher, Waltham, MA, USA, 10,131–035), and nourseothricin (Jena Bioscience, Jena, Germany, AB-101L) were chosen for the experiment. Stocks of each antifungal were made in concentrations of 5120 μg/mL and 7680 μg/mL. Five biological replicates of antifungal concentration were made by pipetting 200 μL of an antifungal into three wells of column 1. A multichannel pipette was used to perform a serial dilution to column 10. At column 10, 100 μL of the culture and antibiotic mixture was pipetted and discarded, leaving 100 μL of mixture in every well. The 96-well plates were incubated at 21 °C for 3 days. The OD600 was measured using Biotek’s Gen 5 software on a BioTek Synergy H1 plate reader (Agilent, Lexington, MA, USA, SH1FSN) with column 12 as a blank. Survival percentage was calculated by the following equation: \(\frac_A}_PC}\bullet 100\%\) where OD600A represents the average OD600 associated with a certain antibiotic concentration and OD600PC represents the average OD600 for the positive controls in column 11. Outliers were calculated using an interquartile range and were excluded in survival percent calculations. Outliers were distributed nearly evenly across all four antibiotics treatments with 16 in the hygromycin data, 13 in the nourseothricin data, 15 in the zeocin data, and 20 in the geneticin data. Outliers were calculated and removed due to imperfect measurements of plate readers with heavy yeast cells, such as X. dendrorhous. Data calculations and figures were made with Microsoft Excel (https://office.microsoft.com/excel).
X. dendrorhous transformation
Integrative pathway DNA for transformation was excised from level 2 vectors with a BsaI digestion. This digestion was comprised of 40 μL plasmid DNA, 5 μL 10X CutSmart Buffer, 2 μL BsaI, and 3 μL nuclease free water. The reaction was run at 37 °C for 10 h, 80 °C for 20 min, and a hold at 10 °C. The resulting DNA fragments were then purified with the Zymo Clean and Concentrator Kit (Zymo, Irvine, CA, USA, D4004) and were ready for transformation. X. dendrorhous transformations were based on an electroporation method described by Visser et al. (2005). X. dendrorhous was first streaked on a YPD agar plate and grown at 21 °C for approximately 2 days. A single colony was used to inoculate 50-mL YPD media in a 125-mL Erlenmeyer flask. The cells were shaken at 21 °C and 200 rpm for another 2 days. These cells were then used to inoculate 200 mL of YPD in a 1-L Erlenmeyer flask at OD600 = 0.02. The cells were grown at 21 °C and 200 rpm until reaching OD600 = 1.2 (approximately 20 h). From here, the cells were pelleted at 1500 × g for 5 min and resuspended in 25 mL of freshly made 50 mM potassium phosphate buffer (pH = 7.0, Sigma Aldrich, Burlington, MA, USA, P8281-100G and P9791-100G) with 25 mM diothiothreitol (Acros Organics, Geel, Belgium, 426,380,500). The cells were incubated at room temperature for 15 min and then pelleted at 4 °C for 5 min at 1500 × g. The cells were then washed with 25 mL of ice cold STM buffer (pH = 7.5, 270 mM sucrose, Millipore Sigma, Burlington, MA, USA, 1.07651.5000, 10 mM Tris HCl, Alfa Aesar, Haverhill, MA, USA, J67233, 1 mM MgCl2, VWR, Radnor, PA, USA, E525-100ML) and spun down again at 4 °C for 5 min at 1500 × g. This wash step was repeated and followed by resuspension of the pellet in 500 μL of STM buffer. Now electrocompetent, the cells were divided into 60 μL aliquots and kept on ice until electroporation. Then, 10 μL of transforming DNA was mixed with a 60 μL aliquot and transferred to an ice cold 0.2-cm electroporation cuvette (ThermoFisher, Waltham, MA, USA, 21–237-2). Electroporation was performed with a Gene Pulser electroporator (Bio-Rad, Hercules, CA, USA, 1998.018.1, 1998.018.2, and 1998.018.3) at 0.8 kV, 1000 Ohms, and 25 μF. Immediately following the electric pulse, 500 μL of ice cold YPD was added to the cuvette. This mixture was then transferred to 4 mL of YPD in a 14-mL Falcon tube and grown overnight on a rotating drum. The next morning, the cells were spun down for 5 min at 1500 × g, resuspended in 500 mL of filtered water, and spread onto selection plates. The plates were incubated at 21 °C until colonies appeared, which typically occurred in 2–3 days. All strain modifications performed on CBS 6938 can be found in Supplemental Table S4. Microsoft PowerPoint (https://office.microsoft.com/powerpoint) was used to generate protocol figures.
Astaxanthin extraction from wild-type and crtYB knockouts
X. dendrorhous CBS 6938 wild-type and crtYB knockout strains were grown for 3 days in the dark at 21 °C and 200 rpm. Then, 1 mL of culture was taken forward and centrifuged at 10,850 × g for 10 min. The cell pellet was washed with Milli-Q filtered water twice before resuspension in 1 mL of acetone (Sigma Aldrich, Burlington, MA, USA, 34,850-1L). Cell and acetone mixtures were poured into pre-filled tubes of 400-micron zirconium beads (OPS Diagnostics, Lebanon, NJ, USA, PFMB 400–100-34) and then transferred to snap-cap microcentrifuge tubes (Eppendorf, Framingham, MA, USA, 022363743). The Bullet Blender Storm Pro tissue homogenizer (NextAdvance, Troy, NY, USA, BT24M) was used at 12 m/s for 5 min to mechanically lyse the cells. Afterward, tubes were centrifuged for 10,850 × g for 10 min. Then, 200 μL of supernatant was pipetted into a nylon syringeless filter (Cytiva, Marlborough, MA, USA, UN203NPENYL) and transferred to a 2-mL chromatography vial (Agilent, Lexington, MA, USA, 5181–3376) with a glass vial insert (5183–2085) and crimp top cap (Agilent, Lexington, MA, USA, 8010–0051).
Chromatography (UPLC) detection of astaxanthin
Ultra-performance liquid chromatography (UPLC) analysis was performed using a modified method from Bohoyo-Gil et al. (2012). Modifications include an injection volume of 10 μL, usage of a Shimadzu Nexcol C18 column (1.8 µm, 50 mm × 2.1 mm) (Shimadzu, Columbia, MD, USA, 220–91394-03), and analysis on the Nexera Series UPLC (Shimadzu, Columbia, MD, USA; RF-20AXS, RID-20A, SCL-40, DGU-403, DGU-405, CTO-40C, SPD-M40, C-40 LPGE, LC-40D XS, SIL-40C XS). The flow rate was 0.3 mL/min and a gradient method was performed. The mobile phase consisted of water treated formic acid (0.1%) (A) acetonitrile (B), ethyl acetate (C), and methanol (D). The gradient followed: 0.00–4.00 min, 85% A and 15% C; 4.00–4.01 min, 60% A, 20% B, and 20% C with a hold until 9 min; and 9.00–9.01 min, 85% A, 15% D with a hold until 10 min. The UV–vis spectra were recorded between 190 and 600 nm. Peaks were detected at 460 nm. The column temperature was maintained at 28 °C. Sample temperatures were maintained at 4 °C. The retention time of astaxanthin was 0.96 ± 0.02. The Shimadzu LabSolutions software (https://www.ssi.shimadzu.com/products/software-informatics/labsolutions-series/labsolutions-cs) and Microsoft Excel (https://office.microsoft.com/excel) were used for analysis. Adobe Illustrator (https://adobe.com/products/illustrator) was used to edit chromatograms.
Bioluminescence detection assay
LPA cultures were started in 2 mL of YPD media at OD600 = 1 and shaken for 24 h at 200 rpm. X. dendrorhous was exposed to either a UV (λ = 400 nm) or dark (no LED light) condition. Bioluminescence was detected using the Nano-Glo Luciferase Assay System (Promega, Madison, WI, USA, N1110). Assay buffer and assay reagent were mixed in a 50: 1 ratio to create to assay solution. Aliquots of 50 μL of X. dendrorhous cells were mixed with 50 μL assay solution in a 96-well plate (Corning, Corning, NY, USA, 3904). Bioluminescence was measured on a BioTek Synergy H1 plate reader (Agilent, Lexington, MA, USA, SH1FSN) with Biotek’s Gen 5 Software for Imaging and Microscopy (https://www.agilent.com/en/product/cell-analysis/cell-imaging-microscopy/cell-imaging-microscopy-software/biotek-gen5-software-for-imaging-microscopy-1623226). Microsoft Excel (https://office.microsoft.com/excel) was used to generate figures.


Comments (0)