Human Participants
All participants with PD (n = 22, mean age = 63.81 ± 7.69 years, aged 50–75, gender M/F = 12/10) were recruited from the inpatient services at Nanjing Medical University Affiliated Brain Hospital and diagnosed with PD in accordance with the UK Parkinson’s Disease Brain Bank criteria. Matched healthy control participants (n = 21, mean age = 60.29 ± 7.72 years, aged 49–77, gender M/F = 9/12) were recruited at the same time. Participants who had obstructive sleep apnea syndrome, dementia, epilepsy, claustrophobia, or psychiatric disorders were excluded. Before the fMRI scan, all participants underwent comprehensive examinations during 12 h –15 h of the off-state. The clinical study was conducted in accordance with the principles of the Declaration of Helsinki and approved by the Medical Ethics Committee of Nanjing Brain Hospital Affiliated with Nanjing Medical University (2021-KY006). Written informed consent was given by all participants after a complete description of the study. Demographic and clinical data, including age, education, sex, disease duration, and clinical symptoms, were registered on the same day. Levodopa equivalent daily dose (LEDD) was calculated according to our previous research [13, 14].
Experimental Animals
Adult male Sprague-Dawley rats and our established transgenic rat strain [15] expressing Cre recombinase in histidine decarboxylase (HDC, the histamine-synthesizing enzyme) neurons (220 g–250 g) were housed under a 12 h light/dark cycle, constant temperature, and humidity (21 °C–23 °C and 40%–60%) with free access to food and water. HDC-Cre rats were generated using CRISPR/Cas9 technology, as we reported previously [15]. Experimental procedures and animal care were in accordance with the Animal Ethics and Welfare Committee of Nanjing University (registration number IACUC-2005006), and the relevant regulations of the NIH Guide for the Care and Use of Laboratory Animals.
Unilateral 6-OHDA lesions (Sigma, St. Louis, USA) were made to establish a rat model of hemiparkinsonism as we reported previously [11]. Adult rats (220 g–250 g) were placed into a stereotaxic frame (1404, David Kopf Instruments, Tujunga, USA) after anesthesia (sodium pentobarbital, 40 mg/kg, i.p., Sigma). 6-OHDA (4 µL, 2.5 μg/μL, dissolved in saline containing 0.02% ascorbic acid; Sigma) was microinjected into the medial forebrain bundle (in mm: A − 4.4, L 1.8, and H 7.8) according to the rat brain atlas of Paxinos and Watson (2013) 30 min after treatment with desipramine hydrochloride (25 mg/kg, i.p.; Sigma) to protect noradrenergic neurons. Two weeks after surgery, the rats were injected with apomorphine (0.25 mg/kg, Sigma) and tested for rotational behavior. Only rats with significant turning behavior (> 25 turns in 5 min) contralateral to the lesion side were considered Parkinsonian rats and retained for further studies. At the end of the studies, the bilateral substantia nigra (SN) was immunostained for tyrosine hydroxylase (TH) to confirm ipsilesional dopaminergic depletion.
Resting-State fMRI in Human Subjects
Resting-state fMRI was acquired in a 3.0 T MRI system (Siemens, Verio, Germany) equipped with a standard 8-channel head coil after anti-PD medications were discontinued for at least 12 h (i.e., during the off-stage) to decrease the medication’s influence on the images as we have reported previously [13, 16]. Subjects were instructed to lie flat, close their eyes, and remain awake in the process of scanning. To restrict head movement, sponge pads were placed on all subjects. T1 images were acquired using the 3D magnetization-prepared rapid gradient-echo (3D-MPRAGE) sequence with the following parameters: repetition time (TR) = 1900 ms, echo time (TE) = 2.48 ms, flip angle (FA) = 9°, matrix size = 256 × 256, field of view (FoV) = 250 mm × 250 mm, slice number = 176, slice thickness = 1 mm, and slice gap = 0 mm. The total scan time was 4 min 18 s. Functional images were obtained using an echo-planar imaging (EPI) sequence (TR = 2000 ms, TE = 25 ms, FA = 90, matrix size = 64 × 64, FOV = 240 mm × 240 mm, slice number = 33, slice thickness = 4 mm, and slice gap = 0 mm). The total scan time was 8 min 6 s.
As we reported previously [17], resting-state fMRI images were all preprocessed using SPM12 (https://www.fil.ion.ucl.ac.uk/spm/) and DPABI (http://rfmri.org/DPABI). The first 10 TRs were discarded to allow magnetization to reach a study state. Slice time correction and head motion correction were then processed. By using the EPI template in SPM12, the functional images were normalized to Montreal Neurological Institute (MNI) space, resampled to 3-mm voxels, and smoothed via a Gaussian kernel with a 6-mm full-width at half-maximum. Then, we applied linear detrending. Moreover, several confounding covariates, including the Friston-24 head motion parameters, white matter, and cerebrospinal fluid, were regressed from the BOLD time series for all voxels. We used Automated Anatomical Labelling Atlas 3 [18] to identify the VA nucleus as in previous studies [19, 20]. Finally, the amplitude of low-frequency fluctuation (ALFF) was calculated to measure brain activity. All the individual whole-brain ALFFs were standardized with the z-score. Then, the independent two-sample t-test was applied to the two groups under the mask of the VA nucleus, which was extracted from the atlas of the adult human brain. Finally, Generalized Ransom Forest correction was applied to the t-test result.
Quantitative Reverse Transcription PCR (qRT‑PCR)
As we have reported previously [11], 5 independent groups of RNA pools, each from 3 animals, were used as biological replicates. VA tissue punches were collected from coronal brain slices from Sprague-Dawley rats (weighing 220 g–250 g) according to the atlas of Paxinos and Watson (2007) and pooled (3 animals in each pool). RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. A 1 μg aliquot of total RNA was used for the first-strand cDNA synthesis according to the protocol of HiScript Reverse transcriptase (Vazyme, Nanjing, CN). Real-time PCR was then applied using qPCR SYBR Green Master Mix (Yeasen, Shanghai, CN) in 20 μL of reaction mixture containing 10 μL of qPCR SYBR Green Master Mix, 2 μL of cDNA, 0.4 μL of each primer (10 μmol/L), and 7.2 μL of distilled water. The reaction was carried out in a Roche light cycler 480 real-time PCR system using the following parameters: 95 °C for 30 s to activate the hot-start iTaq DNA polymerase, followed by 40 cycles at 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 20 s. The PCR program was completed by a melting temperature analysis. The quantity of the target gene was expressed relative to the amount of the reference gene (gapdh) to obtain a normalized target expression value. For negative controls, cDNA was replaced with water. Primer sequences are as follows: mRNAs of GAPDH (forward: TTCAACGGCACAGTCAAGG, reverse: CTCAGCACCAGCAT-CACC), HRH1 (forward: CACTTGAACCGAGAGCGGAA, reverse: CGTGGAGTTG-ATGTAGCCCA), and HRH2 (forward: ATTCGTTTACCGTGGGCTGA, reverse: GAGAGTTGTGGCT-TGCGAAC).
Western Blot
Western blot assays were performed as previously reported [11]. The VA tissue punches were homogenized in 500 μL cell lysate (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1% Triton X-100, 1 mmol/L PMSF, 1% sodium deoxycholate, 0.1% SDS, 2.0 mmol/L EDTA, 10 μg/mL leupeptin). The protein concentration was assessed using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, USA), followed by heating at 95 °C for 5 min. Caudate-putamen (Cpu) and lateral vestibular nucleus (LVN) tissue punches were also performed with the same treatments. The protein samples were separated using 10% SDS polyacrylamide gel, and the proteins in the gels were transferred onto nitrocellulose membranes, which were then placed in a 5% blocking agent for 1 h. The immunoblots were incubated overnight at 4 °C in the primary antibody including mouse anti-GAPDH (1:2000; Santa Cruz, Dallas, USA, Cat# SC-365062, RRID: AB_10847862), rabbit anti-HRH1 (1:200; Alomone Labs, Jerusalem, Israel, Cat# AHR001, RRID: AB_2039915), and goat anti-HRH2 (2 µg/mL; Everest Biotech, Upper Heyford, UK, Cat# EB06905, RRID: AB_2121375), and incubated at room temperature for 3 h in the secondary antibody including horseradish peroxidase (HRP)-conjugated anti-mouse antibody (1:5000; Thermo Fisher Scientific, Cat# 62-6520, RRID: AB_88369), HRP-conjugated anti-rabbit antibody (1:5000; Thermo Fisher Scientific, Cat# 62-6520, RRID: AB_88369), and HRP-conjugated anti-goat antibody (1:5000; Thermo Fisher Scientific, Cat# 31402, RRID: AB_228395). After washing with TBST (tris-buffered saline containing 0.2% Tween 20), the protein entity complexes were visualized by the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) and exposed to Kodak medical X-ray film (Denville Scientific Inc., Holliston, USA).
Immunofluorescence Staining
Immunostaining was applied as in our previous reports [15, 17, 21]. Rats of either sex were deeply anesthetized with sodium pentobarbital and transcardially perfused with 100 mL saline, followed closely by 250 mL–300 mL 4% paraformaldehyde or 4% N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDAC; Sigma; for histamine immunostaining) in 0.1 mol/L phosphate buffer. Brains were immediately removed and postfixed in 4% paraformaldehyde at 4 °C overnight or 4% EDAC for 4 h followed by 4% paraformaldehyde at 4 °C overnight. Each brain was then dehydrated and cryoprotected by incubation in 20% sucrose, then 30% sucrose, for 24 h each. The frozen brain was cut into 25-μm-thick coronal sections containing the TMN, VA, and SN, which were stabilized on gelatin-coated slides by a freezing microtome (CM3050S, Leica, Germany). Sections were rinsed extensively in PBS containing 0.1% Triton X-100 (PBST) and then blocked in 10% normal bovine serum in PBST for 30 min.
The sections were incubated at 4 °C overnight with primary antibodies against tyrosine hydroxylase (rabbit anti-tyrosine hydroxylase antibody, 1:500; Abcam, Cambridge, UK, Cat# AB137869, RRID: AB_2801410), histidine decarboxylase (rabbit anti-histidine decarboxylase antibody, 1:200; Progen, Wayne, USA, Cat# 16045, RRID: AB_2773044), NeuN (rabbit anti-NeuN antibody, 1:500; Proteintech Group, St. Rosemont, USA, Cat# 26975-1-AP, RRID: AB_2880708), H1 receptor (rabbit anti-histamine H1 receptor antibody, 1:500; Alomone Labs, Cat# AHR-001, RRID: AB_2039915), and H2 receptor (goat anti-histamine H2 receptor antibody, 1:500; Everest Biotech, Cat# EB06905, RRID: AB_2121375). The sections were washed with PBS after each step and followed by the related secondary antibodies (1:2000; Invitrogen) conjugated to AlexaFluor 647, AlexaFluor 555, and AlexaFluor 488 for 2 h in the dark at room temperature. All high-resolution images were captured using a laser confocal microscope (LSM 980 with Airyscan 2; Zeiss, Oberkochen, Germany). The number of dopaminergic neurons in the SN was estimated by counting the number of neurons within 3D optical dissectors that were extensively spaced at random. Ten optical dissectors sized 100 μm × 100 μm × 50 μm were randomly examined, and the number of positive cells was quantified using the principle of the optical dissector. The density of cells was estimated using the following formula: Nv = Q/v (dis), where Q is the average number of cells counted per dissector, and v (dis) is the volume of the dissector: v (dis) = a [frame] × h, where a is the area of frame and h is dissector height. Data are presented as the number of cells per cubic millimeter.
Whole-Cell Patch Clamp Recording
Brain slices containing the VA were cut at 300 μm on a vibratome (VT 1200S, Leica). Whole-cell patch-clamp recordings were performed on VA neurons in brain slices to assess the receptor and ionic mechanisms. The artificial cerebrospinal fluid used for whole-cell patch clamp recordings was as follows (in mmol/L): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 26 NaHCO3, 2 CaCl2, and 20 D-glucose, and was equilibrated with 95% O2 and 5% CO2. Whole-cell patch-clamp recordings were conducted using glass micropipettes (3 MΩ–5 MΩ) filled with an internal solution of (in mmol/L) 140 K-methyl sulfate, 7 KCl, 2 MgCl2, 10 HEPES, 0.1 EDTA, 4 Na2-ATP, and 0.4 GTP-Tris, adjusted to pH 7.25 with 1 mol/L KOH. The VA neurons were visualized under an Olympus BX51W1 microscope (Olympus, Tokyo, Japan) equipped with infrared differential interference contrast. An Axopatch-700B amplifier (Axon Instruments, Foster City, USA) was used for whole-cell patch-clamp recordings. The signals were fed into a computer through a Digidata-1550 interface (Axon Instruments) for data capture and analysis (pClamp 10.0, Axon Instruments).
The dosages of drugs we applied were in line with our previous reports [13, 22, 23]. Before bath application, the whole-cell current of a neuron was recorded for at least 10 min to assure stability. Then histamine (30 μmol/L–300 μmol/L, Tocris, Bristol, UK) was administered by bath application (1 min). At least 20 min was allowed for cells to recover after drug application to avoid desensitization. Whether the effect of histamine was postsynaptic was assessed by combined application of tetrodotoxin (TTX; 0.3 μmol/L, Alomone Labs), the selective AMPA receptor antagonist NBQX (10 μmol/L, Tocris), the selective NMDA receptor antagonist D-AP5 (10 μmol/L, Tocris), and the selective GABAA receptor antagonist SR 95531 (10 μmol/L, Tocris). Selective agonists for the histamine H1 receptor (2-PyEA, 300 μmol/L, Tocris) and H2 receptor (dimaprit, 300 μmol/L, Tocris), as well as selective antagonists for the H1 receptor (mepyramine, 3 μmol/L, Tocris) and the H2 receptor (ranitidine, 3 μmol/L, Tocris) were applied to examine the underlying postsynaptic receptor mechanism. To characterize the histamine-induced whole-cell current, in voltage-clamp recording, current-voltage plots (I–V curves) were constructed before and during activation of histamine H1/H2 receptors with 2-PyEA/dimaprit using a slow ramp command (dV/dt = − 10 mV/s, ranging from − 60 mV to − 120 mV) to allow the attainment of steady-state conditions. To verify the coupled channels underlying H1 and H2 receptors, ZD7288 (50 μmol/L, Tocris), Tertiapin-Q (100 nmol/L, Tocris), and apamin (100 nmol/L, Tocris) were applied. Moreover, a direct 50-pA hyperpolarizing current was injected to determine the impact of histamine and the ion channels coupled to histamine receptors on the rebound firing of VA neurons.
Stereotaxic Implantation and Microinjection
Male rats (220 g–250 g) were anesthetized with sodium pentobarbital (40 mg/kg) intraperitoneally and then mounted on a stereotaxic frame (1404, David Kopf Instruments) for stereotactic brain surgery under aseptic conditions. A heating pad was used to maintain rectal temperature at 36 °C–38 °C. Briefly, two stainless-steel guide tubes (length 11 mm, o.d. 0.8 mm, i.d. 0.5 mm) for the microinjection cannulae were implanted into the bilateral VAs of each rat. The lower ends of the guide tubes were positioned 2.0 mm above the VA (in mm: A − 2.04, L 1.65, and H 5.8). After surgery, animals were caged individually and allowed to recover for at least 3 days.
For microinjection into the VAs, two injection cannulae (length 13 mm, o.d. 0.5 mm, i.d. 0.3 mm) were inserted to protrude 2 mm beyond the tip of the guide tube. The lower ends of the injection cannulae were just above the bilateral VAs to minimize lesioning of the nuclei. Saline (0.9% NaCl), selective antagonists for the H1 receptor (mepyramine, 3.5 μg, Tocris), and H2 receptor (ranitidine, 4 μg, Tocris) were microinjected with Hamilton syringes (0.5 µL per side, lasting 2 min). Based on our previous studies [11, 17, 24, 25], the effective diffusion ranges of drugs were determined by extracellular electrophysiological recording, and the injection drug doses in this study were tested to ensure that the diffusion areas of drugs were limited to the VA. Data from rats in which implantation sites were histologically identified to deviate from the target brain regions were excluded from further analysis.
Optogenetic Manipulation
For virus injections, VA (in mm: A − 2.04, L 1.65, and H 5.8) or TMN (A − 4.5, L 1.2, and H 9.6) was infused bilaterally with 0.4 µL of rAAV2/9-hSyn-hChR2-mCherry (Taitool, Shanghai, China) or rAAV2/9-hSyn-DIO-hChR2-EGFP (Taitool) through 33-gauge needles.
As we reported previously [15, 17], the single end of a 2 × 1 fiber splitter (Newdoon, Hangzhou, CN) was connected to a rotating commutator (Doric, Quebec, Canada), which was then attached via a fiber to a laser (Newdoon). Light output was measured with an optical power meter and adjusted to 7 mW of 473-nm light. For photostimulation of VA neurons or TMN-VA histaminergic afferent terminals during the behavioral tests, blue light was applied at a pulse width of 10 ms and a frequency of 10 Hz.
Behavioral TestsOpen-Field Test
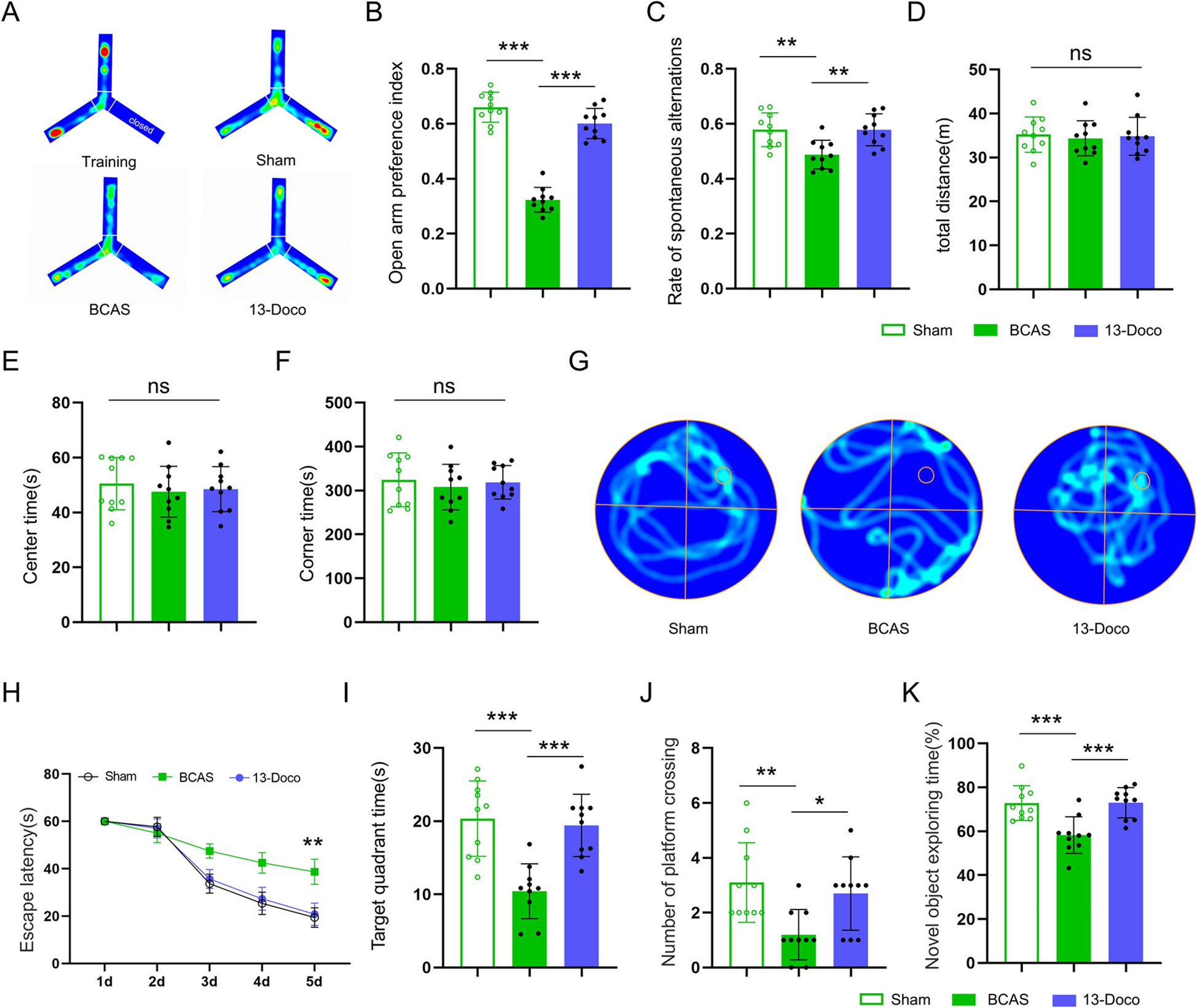
As we have reported previously [11, 15, 24], spontaneous locomotor activity was evaluated by locating individual rats for 10 min in an open-field arena (50 cm × 50 cm × 50 cm) with a video camera positioned at the top of the arena. Rats were placed in the center of the arena at the beginning of the open-field test. Total travel distance and instantaneous velocity were quantified to estimate locomotion. The percentage of time spent in the central zone (25 cm × 25 cm) was used to measure anxiety.
Rotarod Test
As in our previous reports [11, 15, 24], an accelerating rotarod test was applied to estimate motor coordination and balance. For this test, rats were subjected to 3 trials, with 5 min inter-trial intervals to eliminate fatigue and stress. The average latency to fall from the rod in a session (3 repeat trials) or cling to and rotating with the rod was recorded to assess rotarod performance. Rats were adapted to the treadmill (Model 47650, Ugo Basile, Varese, Italy) before the accelerating rotarod test and were required to keep walking and avoid dropping from the rod for 15 s. When testing, the rotation speed was initially set at 5 r/min for 15 s, and then increased to 40 r/min for 300 s.
Turning Behavior Test
As we have reported previously [11], the 6-OHDA-lesioned rats were given an intraperitoneal injection of apomorphine (0.25 mg/kg, i.p.) 3 weeks after surgery. The number of rotations contralateral to the lesion was counted 30 min after apomorphine injection.
Adhesive-Removal Test
As we have reported previously [11], the adhesive-removal test was applied to assess motor initiation and execution. Two training trials were conducted by placing two adhesive tapes (8 mm × 6 mm) on the plantar surface of both forelimbs simultaneously. The rat may bring one forelimb onto the other forelimb and use its mouth to remove the adhesives. We trained the animals for 5 days (1 trial per animal per day; each trial took a maximum of 3 min). On day 6, the time for PD rats to remove the tape was recorded. If a rat failed to remove either or both stickers within 60 s, a score of 60 s was recorded.
Wire-Hanging Test
As has been reported previously [26, 27], the wire-hanging test was applied to assess grip strength and coordination. A metallic wire (2 mm in diameter, 60 cm long) was stretched horizontally between 2 vertical stands, 50 cm above the ground, the rat was placed midway and then left for a maximum of 5 min. A foam pad was situated beneath the rat to prevent injury due to falls. Each rat was experimenter-rated according to the following scores: 0, falls off; 1, hangs onto string by two forepaws; 2, as for 1, but attempts to climb to the string; 3, hangs onto the string by two forepaws plus one or both hind paws; 4, hangs onto the string by forepaws with tail wrapped around string; 5, escapes.
Statistical Analysis
All data are expressed as the mean ± SEM and were analyzed using SPSS 17.0 software. Student’s t-test or one-way analysis of variance (ANOVA) was applied to determine the statistical significance of differences. P values < 0.05 were considered to be significantly different.


Comments (0)