
Remember me


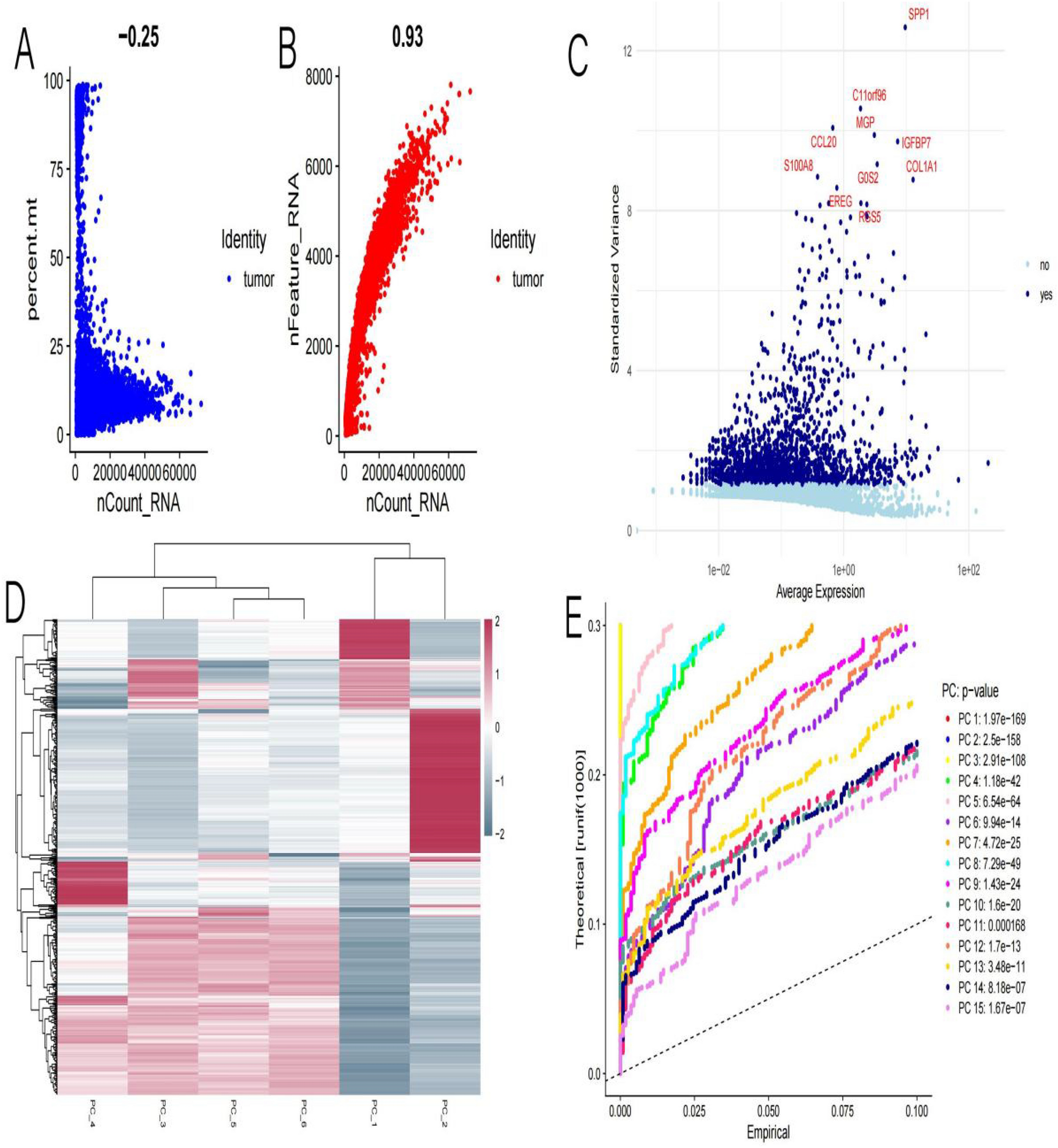
Based on the mRNA-Seq data of 88 COVID-19 patients in the GEO database (GSE171110, GSE152418), and 209 pancreatic cancer patients in GSE15471 and GSE62165, we screened differentially expressed genes separately. We showed that 1201 genes were significantly differentially expressed in COVID-19 patients, including 757 upregulated genes (logFC ≥ 1, P < 0.05). In PC, there were 745 genes that were significantly differentially expressed, including 515 upregulated proteins (logFC ≥ 1, P < 0.05). The heat map (Fig. 2A, B) and volcano map (Fig. 2C, D) of differentially expressed genes for the two diseases were plotted separately. Then, we performed an intersection of the upregulated genes in both diseases, and the results showed that 73 proteins were significantly upregulated in both diseases (Fig. 2E). We then analyzed the functions of the 73 significantly upregulated proteins and evaluated the major diseases in which they were enriched. The results indicated that these genes were mainly enriched in functional areas such as mitochondrial cell cycle, angiogenesis, enzyme-linked receptor protein signaling pathway, regulation of vasculature development, positive regulation of chemotaxis, circulatory system process, mitotic cytokinesis, rhythmic process, and others. The WikiPathways were mainly enriched in the network map of the SARS-CoV-2 signaling pathway, retinoblastoma gene in cancer, and other pathways (Fig. 2F, G). Additionally, the disease ontology (DO) enrichment analysis showed that these proteins are primarily involved in recurrent tumors, pancreatic tumors, adenoid cystic carcinoma, malignant mesothelioma, and other diseases (Fig. 2H).
Fig. 2
Differentially expressed gene screening and functional/pathway analysis. A, B Heat map of differentially expressed gene screening for two diseases, sequentially COVID-19, PC. C, D Volcano map of differentially expressed genes, sequentially COVID-19, PC. E Wayne plot of the intersection of up-regulated proteins expressed in both diseases. F, G Functional and pathway analysis, F is a bar graph and G is a network relationship graph. H Bar graph of disease-specific enrichment analysis
3.2 Screening for genes with common characteristics of COVID-19 and PCWe then further screened 73 intersecting up-regulated proteins in both diseases. Based on the expression levels of upregulated proteins in PC, we first performed a machine learning screening of the included proteins using a support vector machine (SVM-RFE. Setting parameters: seed = 123; Ganna = 0.1; Cost = 1; Accuracy: 0.708), which showed minimal cross-validation when the result was 34, thus screening out 34 upregulated proteins (Fig. 3A). Next, we further analyzed the 73 included proteins using LASSO-Cox regression analysis(Setting parameters: seed = 123; Nfolds = 10), which showed that 14 proteins were screened out when the cross-validation result was minimal (Fig. 3B). Then we again intersected the results screened by the two different machine learning methods, and the results showed that 8 proteins were selected in both machine learning methods (Fig. 3C). To ensure the credibility of the results, we again analyzed the screened intersection genes using the random forest tree learning method and ranked the importance of the 8 included proteins, then selected the top 3 proteins for subsequent analysis. The results showed that the RF Ntree (Setting parameters: seed = 123; Ntree = 500; RfGenes=[1:3]) took the minimum error at position 72, and the importance bubble plot showed that COL10A1/FAP/FN1 were the top 3 proteins in terms of importance, respectively (Fig. 3D, E). Then, COL10A1/FAP/FN1 were defined as the common signature genes of COVID-19 and PC in this study.
Next, we further determined the expression levels of the three signature genes based on the expression levels of COL10A1/FAP/FN1 in PC, and the results showed that the expression levels of COL10A1/FAP/FN1 were significantly upregulated in dataset GSE15471 (Fig. 3F–H, logFC > 1.5, P < 0.001), and the results were validated using dataset GSE62165, which also showed that the expression of COL10A1/FAP/FN1 was significantly upregulated compared to the paracancerous tissue (Fig. 3I–K, logFC > 1.5, P < 0.001). In this study, the diagnostic efficacy of the three signature genes for PC was also analyzed, and the results showed good diagnostic efficacy of COL10A1/FAP/FN1 for PC (ROC-AUC > 0.9) in both datasets GSE15471 (Fig. 3L–N) and GSE62165 (Fig. 3O–Q).
Fig. 3
Feature gene screening. A Support vector machine(SVM) screening of feature genes. B LASSO-Cox regression analysis to screen the feature genes. C Venn diagram of the intersection of support vector machine(SVM) and LASSO-Cox regression analysis screening results. D, E Random forest tree (RF) plots of the intersection results and their corresponding importance ranking bubble plots. F–H Differential expression box plots of COL10A1/FAP/FN1 in dataset GSE15471. I–K Differential expression box plots of COL10A1/FAP/FN1 in dataset GSE62165. L–N Diagnostic ROC curves of COL10A1/FAP/FN1 on PC in dataset GSE15471. O–Q Diagnostic ROC curves of COL10A1/FAP/FN1 against PC in dataset GSE62165
3.3 Construction and validation of the nomogram risk modelWe found that the expression levels of COL10A1/FAP/FN1 were significantly upregulated in PC and showed good diagnostic efficacy for PC, suggesting that these three proteins are diagnostic targets for PC. Based on the expression levels of COL10A1/FAP/FN1, we further constructed a nomogram risk model for pancreatic cancer. From this risk model, we can see that the upregulation of COL10A1/FAP/FN1 expression is a risk factor for pancreatic cancer, and the upregulation of all three increases the risk of developing PC in patients (Fig. 4A). Then, to verify the efficacy of the model, we plotted the calibration curves, which calculates the predicted probability for each observation based on the model, divides them into 10 groups based on increasing predicted probabilities, and then calculates the incidence rate for each group by comparing the predicted outcomes from the model with the actual observed outcomes. The scatter plot is created by plotting the corresponding incidence rates on the y-axis against the predicted probabilities on the x-axis. The results showed that the calibration curves displayed a good fit, suggesting that the risk model has good predictive efficacy (Fig. 4B). The clinical applicability of the risk model was then analyzed, and the decision curve (DCA) showed a good net clinical benefit (Fig. 4C), and the positive detection rate of the model in the clinical impact curve was also very close to the number of real positive patients, further supporting the predictive effectiveness of the model (Fig. 4D). Taken together, the PC risk model based on the characteristic genes COL10A1/FAP/FN1 has good predictive efficacy and clinical application potential.
Fig. 4
Construction and validation of risk models. A The nomogram risk model is based on COL10A1/FAP/FN1 expression. B Calibration curve. C Clinical decision curve (DCA) of the model. D Clinical impact curves of the model
3.4 Subtyping of pancreatic cancer and its correlation analysisNext, in this study, PC was subtyped using unsupervised cluster analysis based on the expression levels of COL10A1/FAP/FN1. The results showed that the included samples effectively scored into two subtypes, C1 and C2 (Fig. 5A–C). To verify the validity of the typing, we also performed principal component analysis (PCA) on the included samples. The results demonstrated that the samples were clearly divided into two groups, A and B, which was consistent with the cluster analysis results, confirming the validity of the typing (Fig. 5D). Furthermore, we analyzed the expression of the three characteristic genes between the two subtypes. The results revealed that COL10A1/FAP/FN1 were highly expressed in the C1 subtype and less expressed in the C2 subtype. All three genes exhibited the same expression trend between the two subtypes (Fig. 5E). We also investigated the survival differences between the two subtypes. The results showed a significant difference in survival between the C1 and C2 subtypes, P = 0.35. (P < 0.05), with the C1 group associated with a poorer prognosis and the C2 group with a better prognosis (Fig. 5F). In addition, we analyzed the mutation of common tumor mutation genetic loci between the CI and C2 subtypes. The results revealed that the mutation rate between both subtypes was over 85%. Furthermore, several mutation loci such as TP53, TNX8, TGFBR2, MYO16, and CACNA18 displayed significantly higher mutation rates in C1 (signature gene high expression group) compared to C2 (signature gene low expression group). This suggests that C1 has higher mutation rates at certain mutant loci (Fig. 5G, H). We then evaluated the tumor microenvironment of the two subtypes. The results indicated significant differences in the immune score, stromal score, and overall score of the tumor microenvironment between the C1 and C2 subtypes, indicating marked dissimilarities in the tumor immune microenvironment (Fig. 5I–K, P < 0.001). Based on these results, we further analyzed the expression levels of HLA factors between the two subtypes. The results demonstrated remarkable differences in the expression of HLA factors, such as HLA-A, HLA-DMA, HLA-DMB, HLA-DOA, HLA-DOB, HLA-DPA1, HLA-DPB1, HLA-DPB2, HLA-DQA1, HLA-DQA2, HLA-DQB1, HLA-DQB2, HLA-DRA, HLA-DRB1, HLA-DRB5, HLA-DRB6, HLA-H, and HLA-L, between C1 and C2 (Fig. 5L, P < 0.005). Additionally, we analyzed the expression of common immune checkpoints between the two subtypes. The outcomes revealed marked differences in the expression of various immune checkpoints, including classical immune checkpoints such as CD274, PDCD1, and CTLA4 (Fig. 5M, P < 0.001).
Fig. 5
Signature gene-based pancreatic cancer typing and its correlation analysis. A–C Results of unsupervised cluster analysis in PC based on COL10A1/FAP/FN1 expression levels. D Principal component analysis (PCA) scatter plots to validate the clustering analysis results. E Box plots of expression analysis of the signature genes in the two subtypes. F Prognostic analysis between the two subtypes. G Mutation status of common tumor mutation loci in C1 subtypes. H Mutation status of common tumor mutation loci in C1 subtypes. I–K Violin plots of the results of tumor microenvironment analysis between the two subtypes. L Box plot of leukocyte factor expression between the two subtypes. M A box plot of the expression of common immune checkpoints between the two subtypes
3.5 Tumor immune correlation analysisNext, the tumor microenvironment of the entire pancreatic cancer (PC) was analyzed in this study. We compared the composition of each immune cell in the tumor microenvironment between paraneoplastic tissues and PC (Fig. 6A), as well as the relationship between them (Fig. 6B). The results revealed that the levels of TFH, resting NK cells, monocytes, macrophages M0, macrophages M1, macrophages M2, resting mast cells, and activated mast cells were remarkably different in paraneoplastic tissues and PC. The expression levels of TFH, resting NK cells, monocytes, and resting mast cells in PC were significantly lower than those in paraneoplastic tissues, while the aggregation levels of macrophages M0, macrophages M1, macrophages M2, and activated mast cells in PC were higher than those in paraneoplastic tissues, and there was a negative correlation among most tumor immune cells (Fig. 6C, P < 0.05). We then analyzed the correlation between COL10A1/FAP/FN1 and tumor immune cells, and the outcomes showed that FAP had significant positive correlations with macrophages, Th1 cells, neutrophils, iDC, Tgd, TReg, mast cells, and NK cells (Fig. 6D, R > 0.4, P < 0.001). COL10A1 showed significant positive correlations with macrophages, Th1 cells, and neutrophils (Fig. 6E, R > 0.4, P < 0.001). FN1 mainly showed a significant positive correlation only with Th1 cells (Fig. 6F, R > 0.4, P < 0.001). In combination with the analysis results, COL10A1/FAP/FN1 were mainly positively correlated with most of the tumor immune cells, which might be associated with an enhanced tumor immune response. Based on the previous significant differences in the expression levels of the two subtypes of C1 and C2 immune checkpoints, we further investigated the correlation between COL10A1/FAP/FN1 and tumor immune checkpoints. The results showed that FAP had significant positive correlations with CD276, CD86, CD200, CD44, CD70, PDCD1LG2, HAVCR2, CD200R1, CD27, TNFRSF8, CD48, CD40, TNFRSF9, NRP1, CD80, CD28, ICOS, LAIR1, TIGIT, CTLA4, TNFSF4, CD274, and other immune checkpoints (Fig. 6G, R > 0.4). COL10A1 also showed marked positive correlations with some immune checkpoints, such as TNFSF9, CD276, CD86, CD44, CD70, LGALS9, PDCD1LG2, HAVCR2, CD40, TNFRSF9, NRP1, CD80, LAIR1, HHLA2, and TNFSF4 (Fig. 6H, R > 0.4). FN1 mainly showed significant positive correlations mainly with TNFSF9, CD276, CD86, CD44, CD70, PDCD1LG2, HAVCR2, CD40, TNFRSF9, NRP1, CD80, LAIR1, TNFSF4, and CD274 (Fig. 6I, R > 0.4).
Fig. 6
Tumor immune correlation analysis. A Bar graph of the composition of tumor immune cells in paracancerous tissue and PC. B Heat map of the correlation analysis between each tumor immune cell. C Violin plot of the difference analysis between the levels of tumor immune cells in paraneoplastic tissues and PC. D–F Bar graphs of correlation analysis between FAP/COL10A1/FN1 and each tumor immune cell in order. G–I Heat map of correlation analysis between FAP/COL10A1/FN1 and each immune checkpoint, in order
3.6 Signaling pathway analysis of FN1This study also investigated the signaling pathways involved in COL10A1/FAP/FN1, and the results showed that FN1 is a component of the extracellular matrix (ECM), which is a characteristic gene for cancer. The ECM components and factors can activate the PI3K-AKT signaling pathway through integrins ITGA and ITGB, thereby affecting the cell cycle and regulating the signaling process in cancer. This process is closely related to the proliferation and apoptosis of cancer cells. Based on this, we hypothesize that the upregulation of FN1 expression in PC may promote the progression of pancreatic cancer and is associated with a poorer prognosis for patients (Fig. 7).
Fig. 7
Schematic representation of FN1 activation of PI3K-AKT signaling pathway
3.7 Drug sensitivity analysis between C1 and C2 subtypesAt the end of the study, the IC50 value was calculated based on the pRRophetic algorithm and signature genes expression profile to predict the sensitivity of different chemotherapeutic drugs between C1 and C2 subgroups. The results showed that the C1 subgroup exhibited higher sensitivity to most drugs compared to the C2 subtype (Fig. 8). Taken together, the results of the typing method and drug sensitivity analysis presented in this study may provide more targeted drug therapy for PC patients. However, further clinical trials are needed to demonstrate the feasibility of the study’s results.
Fig. 8
Box plot of the drug sensitivity analysis between C1 and C2 subtypes
Comments (0)