
Remember me
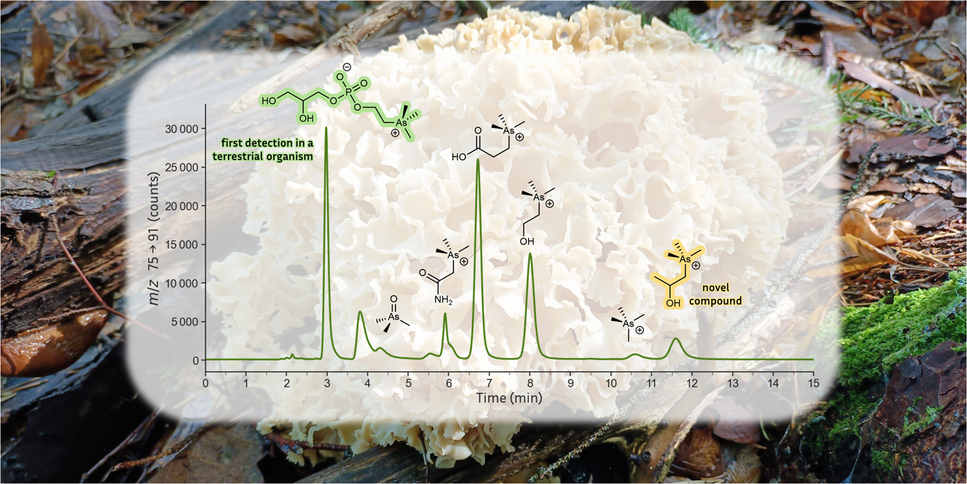
In this study, the fruiting bodies of multiple collections of Sparassis crispa from Austria and the Czech Republic were investigated. Austrian samples (labeled AT1, AT2, and AT3) were collected near Pöllau bei Hartberg (Styria, Austria) and stored in plastic bags during transportation. The fruiting bodies were freeze-dried (Christ, Osterode am Harz, Germany) and homogenized (ultra-centrifugal mill ZM200, 1 mm titanium sieve, Retsch GmbH, Haan, Germany). The Czech sample (CZ1) was collected at Mokrsko village in Central Bohemia, where As content in soil is naturally elevated [19] and was provided as a homogenized powder.
Total arsenic determinationTriplicates of the sample powder (200 mg each) and a certified reference material (CRM) IPE-120 (250 mg, Agaricus bisporus, WEPAL, Wageningen, Netherlands) in 5 mL of HNO3 (p.a., ≥ 65%, Carl Roth, Karlsruhe, Germany, sub-boiled in-house) were subjected to microwave-assisted digestion (Ultraclave IV, MLS GmbH, Leutkirch, Germany; temperature ramp up to 250 °C, pressure up to around 100 bar). Due to the low amount available, a triplicate measurement of the sample from Czechia was not possible; thus, the total As concentration in CZ1 is based on only a single measurement. To the digests, 500 µL of concentrated HCl were added and diluted to a total volume of 50 mL with ultrapure water (18.2 MΩ × cm, Merck Millipore, Bedford, USA). Arsenic was determined by ICPMS (7900, Agilent Technologies, Waldbronn, Germany) or ICPMS/MS (8900, Agilent Technologies, Waldbronn, Germany). The quality of the analysis was confirmed by the aforementioned reference material CRM IPE-120 and the standard reference material SRM® 1643f (Trace elements in water, NIST, Gaithersburg, USA), as well as blanks consisting of ultrapure water and nitric acid added to the autoclave during digestion. The trueness of the measurement was confirmed by comparing the measured to expected arsenic concentration. The deviation was calculated by (cmeasured–cexpect)/cexpected × 100%, arriving at + 1% relative deviation for CRM IPE-120, and –11% for SRM® 1643f, respectively.
Sample extractionArsenic species were extracted by weighing homogenized mushroom samples (200 mg for AT1–AT3; 10 mg for CZ1) in a 15 mL polypropylene tube and adding ultrapure water (4 mL for AT1–AT3; 2 mL for CZ1). The limited amount of sample CZ1 did not allow for use of more material. The tubes were placed in an ultrasonic bath at ambient temperature for 30 min; the phases were separated by centrifugation (15 min at 10,000 g), and the supernatant was heated to 90 °C for 30 min. Following this heating step, additional solids precipitated from solution, which were removed by centrifugation (20 min at 21,000 g) and filtration of the supernatant through a 0.22 µm Nylon syringe filter to give a clear extract. Unheated extracts would become cloudy within 1–2 days of preparation due to the formation of a fine precipitate, leading to potential problems when injecting samples into the HPLC system. Furthermore, the addition of H2O2 to unheated extracts led to vigorous gas development. This was likely due to enzymatic activity, as no gas development was observed when adding H2O2 to heated extracts. Preliminary experiments revealed that heating the supernatant to 90 °C did not have an apparent effect on the arsenic species, and recorded chromatograms were indistinguishable before and after heating. To study the levels of inorganic arsenic, oxidation of potentially present inorganic As3+ to As5+ was achieved by adding 30% H2O2 to a heated extract (1 + 9) and heating the sample to 60 °C for 30 min. To determine the extraction efficiency, 200 µL of the extracts was added to 5 mL of 10% nitric acid and 1% HCl in 5 mL of ultrapure water. These samples were measured alongside the microwave-assisted acid-digested samples when determining the total arsenic content. An extraction efficiency of 98 ± 12% was determined.
Arsenic speciationArsenic species were separated on an Agilent 1290 UHPLC system (Agilent Technologies, Waldbronn, Germany) comprising a binary pump, an autosampler, and a heated column compartment. Chromatographic conditions for cation-exchange and anion-exchange chromatography are summarized in Table 1. For the preparation of the mobile phases, ammonium formate (≥ 99.95% trace metal basis, Sigma-Aldrich) or diammonium hydrogen phosphate (puriss p.a., ACS reagent, ≥ 99.0%, Sigma-Aldrich) was used. The pH of the former was adjusted with formic acid (puriss. p.a. ≥ 98%, Sigma-Aldrich), while the latter was adjusted with 25% aqueous ammonia solution (ultra pure, Chem-Lab). High-resolution mass spectra (HRMS) were obtained from an Agilent ESI-TOF (electrospray ionization time of flight) with a capillary voltage vc = 4000 V, and a nozzle voltage vn = 2000 V. Chromatographic conditions are summarized in Table 1 under the cation-exchange header.
Table 1 Chromatographic conditions employed for the separation of anionic and cationic arsenic speciesSynthesis of arsenic standardsGeneral considerationsAll air-sensitive reactions were carried out under nitrogen using standard Schlenk techniques. AsCl3 was synthesized from As2O3 and SOCl2 according to the literature [20], and then further converted to AsMe3 in a Grignard-type reaction according to the literature [21]. Arsenocholine, 1-bromo-2-propanol, and (2,2-dimethyl-1,3-dioxolane-4)methanol were synthesized in-house.
Synthesis of trimethyl(2-hydroxypropyl)-arsoniumThe novel trimethyl(2-hydroxypropyl)-arsonium cation, or β-methyl arsenocholine (MeAC) as of established trivial naming schemes [22], was synthesized analogous to other arsenocholine homologues by combining trimethylarsine with an equimolar amount of 1-bromo-2-propanol under inert conditions [4, 23]. A detailed description of the procedure is given in the supplementary information. In short, the nucleophilic attack of bromide at propylene oxide gave a mixture of 1-bromo-2-propanol and 2-bromo-propanol in an 80/20 ratio. Addition of trimethylarsine to this mixture gave only a single product, albeit in a low yield of 17%. This is likely due to a combination of the higher proportional presence of 1-bromo-2-propanol and its less sterically demanding electrophilic carbon, which may be attacked more readily.
Found m/z for C6H16AsO: 179.0411; isotope/mass accuracy score: Δppm –0.51 determined by LC-ESI-HRMS.
Synthesis of α-GPACThe phosphate diester α-glycerophosphorylarsenocholine (α-GPAC) was prepared by adjusting a literature procedure for the synthesis of α-glycerophosphorylcholine [24]. As depicted in Scheme 1, to a solution of phosphoryl chloride in methylene chloride, an equimolar amount of 2,2-dimethyl-1,3-dioxolane-4-methanol and excess NEt3 in methylene chloride, followed by an equimolar amount of arsenocholine in pyridine, were added, and the reaction was allowed to run to completion. The reaction mixture was subsequently quenched with water, forming the acetonide-protected α-GPAC (1). The crude, aqueous reaction mixture was subjected to HPLC–ICPMS, which revealed only arsenocholine (25%) and 1 (75%) as the two major arsenic species. Arsenocholine was removed by passing the product solution through Dowex 50 W Hydrogen form cation-exchange resin. The low pH of the resin resulted in the deprotection of the acetonide and the formation of α-GPAC from 1, as evidenced by HPLC–ICPMS. Formation of the product was confirmed by HPLC-ESI-HRMS, where we measured m/z for C8H20AsO6P: 319.0286; isotope/mass accuracy score: Δppm –0.06. Attempted isolation of solid α-GPAC by lyophilization was unsuccessful, as a sticky, brown residue was obtained each time. While the only arsenic species in the product solution prior to lyophilization was α-GPAC (> 98%), notable amounts of arsenocholine and other unidentified arsenic species were detected in the lyophilizate. The final yield of 38% of α-GPAC was therefore calculated from the total As concentration in solution after passing it through the cation-exchange resin to remove any cationic arsenic-containing impurities. Subjecting synthetic α-GPAC to alkaline conditions (0.5% NaOH) at 60 °C for 15 min resulted in its decomposition to the trimethyl-vinyl arsonium ion (TMVA), AC, AB, and inorganic arsenic.
Scheme 1
Synthesis of acetonide-protected α-GPAC (1), and subsequent deprotection under aqueous, acidic conditions. a: 1.) (2,2-Dimethyl-1,3-dioxolan-4-yl)methanol (1.0 equiv.), NEt3 (2 equiv.), DCM, 0 °C, 1 h; 2.) arsenocholine (1.0 equiv.), pyridine, 0 °C to rt, 16 h. b) Dowex 50 W Hydrogen form
Comments (0)