
Remember me
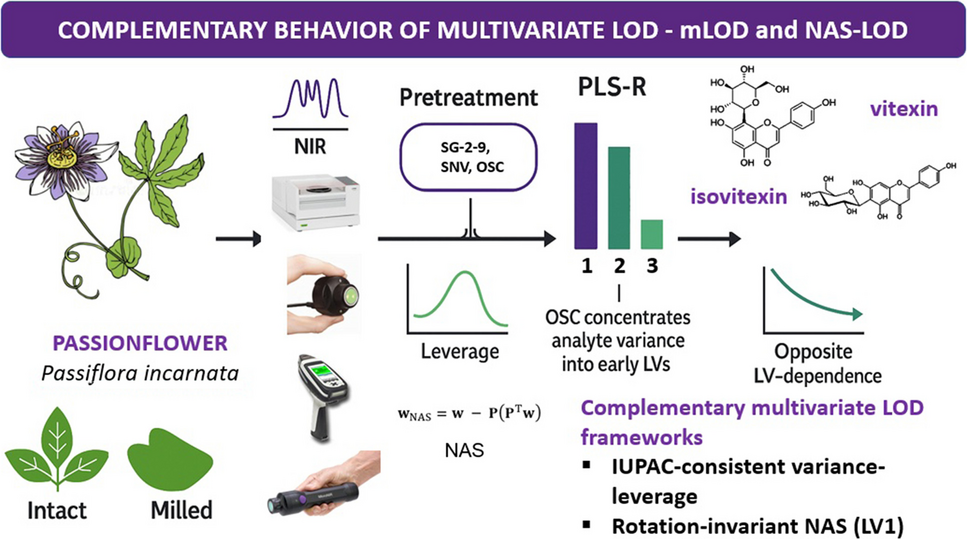
NIR spectra of a representative intact sample measured across all spectrometers are presented in Fig. 1. Despite differences in spectral resolution, wavelength coverage, and optical design, all instruments captured the major absorption features characteristic of phytopharmaceutical matrices. These include broad overtone and combination bands associated with O–H, C–H, and N–H functional groups, typically arising from moisture, carbohydrates, proteins, and other plant constituents. Nonetheless, some distinctions between instruments were observable in the measured lineshape characteristics. Using the benchtop N-500 (with its high stability FT-NIR configuration (section Spectroscopic Measurements)) as the reference, certain contrasting features apparent in the spectra measured by portable LVF-based instruments (the MicroNIR 2200 and 1700 ES) are noticeable. Both these sensors displayed narrower coverage and mild baseline fluctuations, while the 2200 model also featured a rapid decrease in the measured intensity towards the low-frequency boundary (Fig. 1). Yet, even the MicroNIR 2200 retained sufficient spectral fidelity and consistency in critical regions for reliable model construction. On the other hand, the microPHAZIR also reproduced the spectral lineshape of the sample in a specific manner, while featuring the narrowest usable spectral range (6250–4170 cm⁻1) as well, limited to combination band regions. Still, the spectrometer captured informative matrix- and moisture-related features relevant for reliable calibration.
Fig. 1
NIR spectra presented for one representative sample of Passiflora incarnata (intact and milled form) measured using all the spectrometers included in the study. Spectra presented with an arbitrary uniform shift on the intensity axis (vertical offset) for clarity of presentation
PLS regression modelsThe key performance parameters of all developed PLS-R models, constructed separately for each spectrometer, analyte (vitexin, isovitexin, total content), and sample condition (milled/intact), are summarized in Tables 1, 2 and 3. The modelling strategy and evaluation metrics applied are described in the section. As expected, the Büchi NIRFlex N-500 (benchtop FT-NIR) consistently delivered the highest model performance across all analytes and conditions. For milled samples, quantification of vitexin and isovitexin content yielded excellent results, with calibration coefficients of determination (R2C) exceeding 0.99 and prediction errors (RMSEP) typically below 0.5 mg/g and, in the case of vitexin quantification, even below 0.1 mg/g (Tables 1, 2 and 3). These results reflect the upper performance benchmark achievable under controlled laboratory conditions using high-end benchtop instrumentation. The model fit quality for the reference benchtop spectrometer, on the example of intact samples is presented in Fig. 2A–C, for the combined quantification of both analytes (vitexin and isovitexin), and individual ones, accordingly.
Fig. 2
PLS regression line obtained from Büchi NIRFlex N-500 for A vitexin + isovitexin, B isovitexin, C vitexin
In contrast, predictive performance declined when using portable or handheld devices, reflecting inherent instrumental limitations such as reduced spectral resolution, narrower wavelength ranges, and increased noise levels. This trend was most pronounced for the microPHAZIR, which combines narrow spectral coverage with single-channel detection. Additionally, sample condition had a major impact on model quality across all spectrometers. Milled samples consistently produced better predictive results compared to intact material, due to reduced heterogeneity and lower scattering variability, which directly affect NIR spectra and model stability.
Table 1 PLS-R performance metrics for simultaneous quantification of vitexin and isovitexinTable 2 PLS-R performance metrics for quantification of isovitexinTable 3 PLS-R performance metrics for quantification of vitexinSample state further contributed to performance variability; models based on milled samples typically outperformed models based on intact samples, which is a common trend reported in similar studies [39, 40]. This behaviour is attributed to the reduced scattering variability and increased homogeneity achieved by particle size reduction, which suppresses multiplicative and baseline effects that otherwise obscure chemically relevant information in intact samples.
The microPHAZIR, representing the narrowest spectral coverage among the tested instruments, produced the lowest overall model performance. In several cases, test set coefficients of determination (R2TSV) fell below 0.85, and prediction errors approached 0.5 mg/g. This suggests that the quantitative performance was predominantly limited by intrinsic spectral features of the low-frequency NIR range. However, even under these constrained conditions, the resulting models remained quantitatively meaningful, with absolute errors well within the typical good results for quantitative analysis of phytopharmaceutical marker compounds (0.3–10 mg/g). This confirms the suitability of practical in-field deployment of miniaturized NIR instrumentation even in demanding analytical contexts.
Importantly, while Tables 1, 2 and 3 demonstrate the variations in model performance across spectrometers and sample types, they do not directly reveal the actual detection capability of the models in the sense of quantitative LOD or LOQ. Furthermore, these metrics can mask poor analyte alignment, diverging from the PAT principle of structure-transparent and interpretable analytical method. As detailed in the section, the ability of a model to detect and quantify low analyte levels is governed not only by its overall predictive error but by how the analyte-specific variance is represented and isolated within the latent space. This distinction becomes critical when evaluating LOD/LOQ behaviour under different theoretical frameworks.
Comparison of variance-leverage mLOD and NAS-LOD estimatesWhile the calculation of LOD/LOQ represents a fundamental element of quantitative analytical chemistry, its application within multivariate calibration, particularly in phytopharmaceutical products, remains challenging. Tables 4, 5 and 6 systematically compare the two conceptually distinct frameworks for multivariate LOD/LOQ estimation: (i) the variance-leverage mLOD framework and (ii) the NAS-based approach, focusing on the magnitude and delineation of the analyte-specific signal within the latent structure of the PLS-R model.
Both LOD/LOQ estimation frameworks were evaluated using the optimized PLS-R models for vitexin, isovitexin, and their sum (Tables 4, 5 and 6), as quantified across different spectrometers and sample conditions. The modelling was performed using real phytopharmaceutical samples of Passiflora incarnata L., characterized by substantial matrix variability arising from diverse cultivation origins, processing conditions, and inherent physicochemical heterogeneity. The impact of these sample properties on multivariate LOD/LOQ estimation was systematically investigated. A key observation emerging from this study was the divergent behaviour of variance-leverage mLOD and NAS-LOD estimates with increasing model complexity, i.e., with the inclusion of additional LVs. As anticipated, the variance-leverage mLOD values generally improved (decreased) in models with fewer LVs. This reflects the favorable model geometry and reduced calibration variance associated with more compact latent structures, causing reduced average leverage and better separation between the blank and analyte signal within the latent space. Conversely, NAS-LOD values decreased with increasing numbers of LVs, reflecting their direct dependence on the concentration and geometric delineation of analyte-related variance within the latent space. Given that NAS is computed on LV1, the decrease of NAS-LOD with increasing LVs is driven by the drop in CV residual variance. When analyte signal is dispersed across LVs (notably without OSC), NASLV1 is conservative by design, whereas OSC tends to concentrate the analyte in LV1, making NASLV1 both more interpretable and less conservative.
Table 4 Multivariate LOD/LOQ estimates (in mg/g) for vitexin and isovitexin (sum): variance-leverage mLOD and NAS-LODTable 5 Multivariate LOD/LOQ estimates (in mg/g) for isovitexin: variance-leverage mLOD and NAS-LODTable 6 Multivariate LOD/LOQ estimates (in mg/g) for vitexin: variance-leverage mLOD and NAS-LODThis opposite behaviour was consistently observed across spectrometers and analytes, and represents a fundamental consequence of the distinct mechanisms underlying both frameworks. The observed divergence in LOD trends (Tables 4, 5 and 6) very well reflects the difference between the global sensitivity measured by mLOD and the analyte-specific isolation measured by NAS-LOD. The results demonstrate that neither framework should be regarded as universally superior or universally applicable. In matrices with dominant or highly variable background contributions—such as phytopharmaceutical extracts—variance-leverage mLOD may underestimate detection capability if analyte variance is weakly expressed relative to total model variance. Conversely, although not observed in the present results, NAS-LOD values may exhibit overly optimistic behaviour in highly flexible or over-parameterized models, given that a higher number of retained LVs tends to decrease the LOD estimate. This interplay underscores the critical role of model structure and interpretability in multivariate LOD/LOQ estimation. The dependence of NAS-LOD on the geometric separation of analyte-relevant variance is both its advantage (in transparent, well-structured models) and its potential limitation (in unstable or poorly aligned latent spaces). Conversely, the variance-leverage mLOD provides a more global performance estimate, inherently robust to LV rotations, but less sensitive to the true representation of analyte variance within the latent structure.
Particular attention should be given to the limits and interpretative role of multivariate LOD/LOQ conventions. While both frameworks offer valuable perspectives for assessing detection capability in multivariate calibration, their applicability in highly variable phytopharmaceutical systems carries inherent limitations. In complex matrices, increased model flexibility (arising from a higher number of LVs) or elevated calibration variance may lead to reduced separation between low-concentration samples and the calibration cloud in the latent space, resulting in unstable or overly optimistic mLOD estimates that underestimate the actual detection challenges. In contrast, NAS-LOD is directly dependent on the geometric isolation of analyte variance within the model. In cases where the analyte signal is weak, fragmented, or distributed across several LVs—typical for samples with high matrix interference—the resulting NAS vector may become small, yielding conservative, potentially excessively pessimistic LOD estimates, despite adequate external prediction performance.
Behaviour and diagnostic role of LOD/LOQ frameworks in phytopharmaceutical PAT calibrationThe behaviour of LOD and LOQ estimates in PLS-R is fundamentally driven by the internal latent structure of the calibration model, yet the nature and extent of this dependency differ considerably between the variance-based mLOD and NAS-LOD frameworks, as outlined in the section. Two complementary diagnostics allowed systematic quantification of latent structure quality in relation to these LOD frameworks: (i) the variance of the reference analyte explained by each LV, internally evaluating analyte signal compactness or dispersion within the model, and (ii) projection of the pure analyte spectrum onto the model LVs, serving as an external estimate of analyte representation within the latent structure.
Across the considered cases, substantial differences in estimated LOD/LOQ were observed depending on the framework (NAS-LOD vs. variance-based mLOD), model complexity, and preprocessing strategy. Using the N-500 for vitexin and isovitexin quantification, NAS-LOD values below 1 mg/g were achieved consistently in milled samples, slightly increasing in intact samples (Tables 4, 5 and 6). Portable spectrometers, particularly the MicroNIR 2200 and 1700 ES, produced NAS-LOD values generally ranging between 1.5 and 4 mg/g, occasionally exceeding 5 mg/g in intact samples without OSC. Variance-based mLOD estimates exhibited less numerical variability and were less responsive to differences in sample state or preprocessing conditions. Nevertheless, absolute NAS-LOD and mLOD values often converged in well-structured models, suggesting alignment under conditions of optimal analyte delineation. In the case of the microPHAZIR, the LOD values determined through both frameworks led to consistently higher values, particularly for intact samples (Tables 4, 5 and 6), reflecting the intrinsic limitations of spectral range and lower analyte alignment within the latent space.
LV dependence of NAS-LOD versus variance-based mLODA key observation was the divergent behaviour of variance-leverage mLOD and NAS-LOD as the number of retained LVs increased. The values of mLOD improved for more compact models (lower number of LVs), while NAS-LOD computed on LV1 decreased as additional LVs were retained, due to the reduction of the CV residual variance term (the NAS direction remained LV1). For a directly comparable example under identical conditions (same instrument, analyte, sample state, and OSC setting), the N-500, vitexin, intact, non-OSC model shows NAS-LOD decreasing from 13.13 mg g⁻1 (F = 3) to 8.65 mg g⁻1 (F = 5), while mLODavg increases slightly from 3.45 to 3.58 mg g⁻1 (Table 7; N-500, V/i, without OSC). In the corresponding OSC-pretreated model (N-500, vitexin, intact, OSC), NAS-LOD further decreases from 2.94 mg g⁻1 (F = 3) to 1.13 mg g⁻1 (F = 5), accompanied by a minor change in mLODavg from 0.62 to 0.70 mg g⁻1 (Table 7; N-500, V/i, OSC). Together, these controlled examples clearly illustrate the opposite dependence of the two LOD frameworks on model complexity under otherwise identical conditions.
This inverse LV dependence highlights the different roles of the two frameworks. NAS-LODLV1 quantifies the analyte-aligned signal-to-residual contrast in X-space, while the variance-leverage mLOD reflects global predictive sensitivity and leverage-dependent prediction uncertainty. Considering both the analyte-specific delineation (NAS-LOD) and the global detectability conditioned on leverage (mLOD) provides a complementary and more complete model assessment.
Influence of OSC preprocessing on latent structure and LODOSC pretreatment strongly and systematically influenced LOD estimates, predominantly through direct modification of the internal latent structure. The most substantial impact was observed in NAS-LOD values, particularly in intact samples and portable instruments, where background interference was most prominent. OSC-pretreated models consistently achieved meaningful NAS-LOD reductions (Table 7), a direct result of improved analyte signal concentration and isolation within early LVs. The table presents exemplary results for the benchtop and miniaturized spectrometer and varying model complexity (retained LVs) in each case, to assess the stability in the behaviour and potential trends that LOD/LOQ frameworks may follow in a range of method parameters.
Table 7 LOD and LOQ estimates (in mg/g) using variance-based mLOD and NAS-LOD conventions with and without OSC included in the pretreatment schemes. Analysis type: vitexin (V), isovitexin (Iso), intact (i)As anticipated for phytopharmaceutical matrices, the inclusion of OSC in the pretreatment scheme has a significant impact on the predictive power of the models, monitored via routine parameters (R2, RMSE). However, multivariate LOD/LOQ values add considerable depth to the diagnostic layer that provides refined control over the method. A moderate improvement was observed in the isovitexin model using MicroNIR 1700 ES, where mLODavg decreased from 2.52 mg/g (non-OSC) to 2.12 mg/g (OSC). Variance-based mLOD also showed substantial sensitivity to OSC pretreatment in the vitexin model (N-500, intact), where mLODavg decreased significantly from 3.45–3.58 mg/g (non-OSC) to 0.62–0.70 mg/g (OSC), depending on the number of LVs. In this case, OSC reduced mLODavg by factors of approximately 5.6 (3 LVs) and 5.1 (5 LVs), while NAS-LOD dropped by approximately 4.5 and 7.7 times, respectively. These NAS-LOD improvements are consistent with the anticipated higher sensitivity to analyte signal concentration and orthogonality of this multivariate LOD framework.
Additionally, leverage-driven uncertainty narrowed after OSC, as indicated by smaller mLOD min–max intervals (e.g., N-500, vitexin, intact: from 0.17–0.19 mg g⁻1 pre-OSC to 0.05–0.16 mg g⁻1 post-OSC). As the number of retained LVs increased, we observed the characteristic inverse trend; mLODavg rose slightly, whereas NAS-LOD decreased. For the same controlled case (N-500, vitexin, intact, non-OSC), mLODavg increased from 3.45 to 3.58 mg g⁻1 as the number of LVs increased from 3 to 5, while NAS-LOD fell from 13.13 to 8.65 mg g⁻1 (Table 7; N-500, V/i, without OSC). In the corresponding OSC model, NAS-LOD decreased from 2.94 to 1.13 mg g⁻1 (F = 3–5), with mLODavg changing from 0.62 to 0.70 mg g⁻1 (Table 7; N-500, V/i, OSC). These trends highlight the greater structural responsiveness of NAS-LOD to latent space reorganization, while mLOD remained more conservative and less affected by analyte signal fragmentation in the model structure.
Internal diagnostic analysis supported these observations, revealing a marked increase in the proportion of analyte variance captured by LV1 and reduced contributions from higher-order LVs after the application of OSC. This structural shift induced by OSC was quantitatively confirmed by latent variable diagnostics. For the sake of comparison, two examples are provided selected by how the PLS-R latent structure and LOD/LOQ values respond to orthogonal correction. The example selected for the benchtop spectrometer showed a less profound shift, which is also reflected by comparable optimal model complexity (i.e., number of retained LVs) in both OSC and non-OSC scenarios. In contrast, the second example, drawn from models developed using a miniaturized spectrometer, demonstrated a much more pronounced structural reorganization, which substantially altered the optimal model complexity. Accordingly, as shown in Fig. 3A, B, the proportion of analyte variance (vitexin, N-500) explained by LV1 increased markedly in the OSC-pretreated model, while variance in higher LVs diminished. External projection of the pure vitexin spectrum onto the model LVs (Fig. 3C, D) further confirmed this alignment, with the OSC model showing stronger and more focused projection onto LV1. A similar behaviour was observed for isovitexin models based on the MicroNIR 1700 ES (Fig. 4). The analyte variance, originally spread across multiple LVs in the uncorrected model, became sharply concentrated in LV1 (R2 of 0.85) once OSC is included in the pretreatment scheme. This results in a stronger analyte alignment and thus more transparent model structures in the pretreated datasets. Note that the projection was based on the spectrum of a vitexin reference standard, pretreated in a way consistent with the spectra of the natural samples. While such a spectrum — recorded on a pure, polycrystalline substance — cannot fully replicate the analyte signal as it exists within the complex phytopharmaceutical matrix, it provides a chemically grounded external benchmark. Despite these contextual differences, the projection clearly supports the latent structure diagnostics by demonstrating stronger alignment of the standard with LV1 in OSC-pretreated models (Fig. 3C, D). This suggests that the structural reorganization introduced by OSC indeed concentrates analyte-relevant variance on the prime LV and enhances its separation from matrix variance.
Fig. 3
Comparison of PLS-R structure without OSC (A, C) and with OSC (B, D) included in the pretreatment scheme preceding modelling of vitexin (N-500, intact samples). A, B Explained variance of reference analyte per LV, illustrating analyte signal concentration in OSC model (dominance of LV1) versus dispersion across multiple LVs in the non-OSC model. C, D Projection of the pure vitexin standard spectrum onto the latent space (X-loadings), indicating stronger alignment with early LVs in the OSC model
Fig. 4
Comparison of PLS-R structure (presented for 4 LVs) without OSC (A) and with OSC (B) included in the pretreatment scheme preceding modelling of isovitexin (MicroNIR 1700 ES, intact samples). A, B Explained variance of reference analyte per LV, illustrating analyte signal concentration in OSC model (dominance of LV1) versus dispersion across multiple LVs in the non-OSC model
This demonstrates that while NAS-LOD is more directly affected by changes in analyte delineation, variance-based mLOD is not structurally blind. In the example (N-500, vitexin, intact; Table 7), the OSC improves both signal isolation and PLS-R latent structure (i.e., calibration geometry). Although by principle less structurally responsive, mLOD exhibits sensitivity to model reorganization and responds significantly to its realignment. Both NAS-LOD and mLOD provide standardizable diagnostic information on the internal structure of the model, albeit in framework-specific ways. Consequently, both LOD frameworks are structurally responsive and provide complementary, not redundant, diagnostic insights into model quality.
Diagnostic role of LOD behaviour for model structure qualityThese results demonstrate that the LOD frameworks, beyond their quantitative utility for method validation, provide valuable diagnostic insight into model structure and analyte signal integrity. Low NAS-LOD values consistently indicated strong analyte delineation, concentrated variance in early LVs, and high model interpretability—all characteristics in alignment with good-practice PAT expectations. Elevated NAS-LOD, by contrast, reliably exposed models with fragmented or poorly isolated analyte signal, even when conventional error metrics (e.g., RMSEP) or variance-based mLOD suggested acceptable performance. These cases revealed that NAS-LOD responds to geometric analyte alignment within the latent structure, and therefore detects weaknesses not apparent from global error alone. Variance-based mLOD, in contrast, remained less sensitive to such structural dispersion, responding instead to the general geometric relationship between low-concentration samples and the overall latent space constructed in the model. In other words, mLOD does not provide a clear insight into method interpretability. As discussed earlier, this explains why models with similar mLOD but widely differing NAS-LOD may exhibit meaningful differences in analyte interpretability, providing useful metrics for method assessment at a deeper level.
In summary, the combined evaluation of LOD frameworks on the background of spectrometer types (benchtop, miniaturized), analytes (vitexin, isovitexin, joint), and model complexities revealed consistent diagnostic trends. Variance-based mLOD estimates were generally more stable than NAS-LOD across instruments and analytes, but showed substantial responsiveness to structural reorganization in select models. Conversely, NAS-LOD reflects analyte signal concentration and serves as a general indicator of method interpretability. Benchtop spectrometer (N-500) consistently achieved lower detection limits than miniaturized spectrometers, with vitexin models generally outperforming isovitexin, particularly for NAS-LOD. Models quantifying the sum of vitexin and isovitexin exhibited intermediate detection behaviour. Model complexity had opposing effects on the two frameworks; increasing it lowered NAS-LOD but increased mLOD, reflecting the fundamental distinction between analyte-specific variance recovery and global calibration variance. OSC systematically improved both frameworks by compressing model variance and enhancing analyte isolation, with a stronger effect on NAS-LOD. Future studies should focus on developing direct interpretation approaches that link latent space structure with LOD framework-specific detection behaviour, enabling more transparent, standardized calibration diagnostics in phytopharmaceutical applications of NIR spectroscopy.
Comments (0)