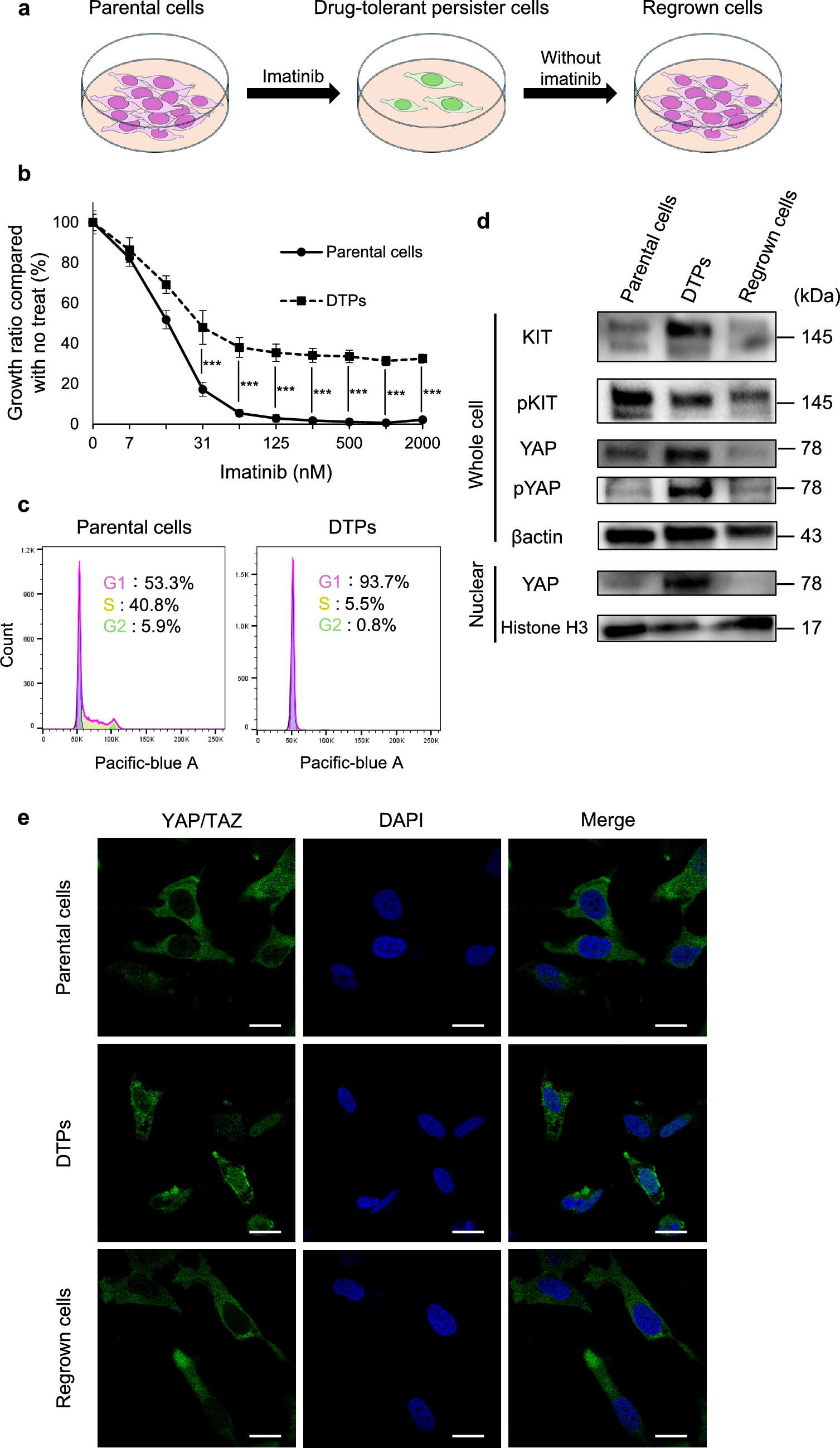
Drug-tolerant persister cells (DTPs) are a subpopulation of cancer cells that survive therapeutic pressure through reversible, non-genetic mechanisms. In contrast to acquired resistance driven by genetic alterations, such as secondary KIT mutations, DTPs evade apoptosis and enter a transient drug-tolerant state without harboring permanent genomic changes [8,9,10]. In our previous work, we demonstrated that secondary KIT mutations were observed only in GIST-T1 cells subjected to long-term imatinib exposure, but not in cells exposed for a short duration [22], supporting the idea that early-phase resistance is mediated by non-genetic adaptations. Consistent with this, the present study revealed that YAP activity was markedly upregulated in DTPs following imatinib treatment, as evidenced by increased nuclear localization. Notably, this change was reversible upon drug withdrawal, further indicating the plastic, non-genetic nature of the DTP state. These findings suggest that GIST cells can evade imatinib-induced apoptosis by dynamically reprogramming their gene expression profiles, with YAP functioning as a key regulator of this adaptive survival response. Although previous reports have described the association between YAP overexpression and malignancy in GIST [16], our study observed the reversible changes in YAP expression in GIST DTPs. This aspect highlights the novelty of our findings.
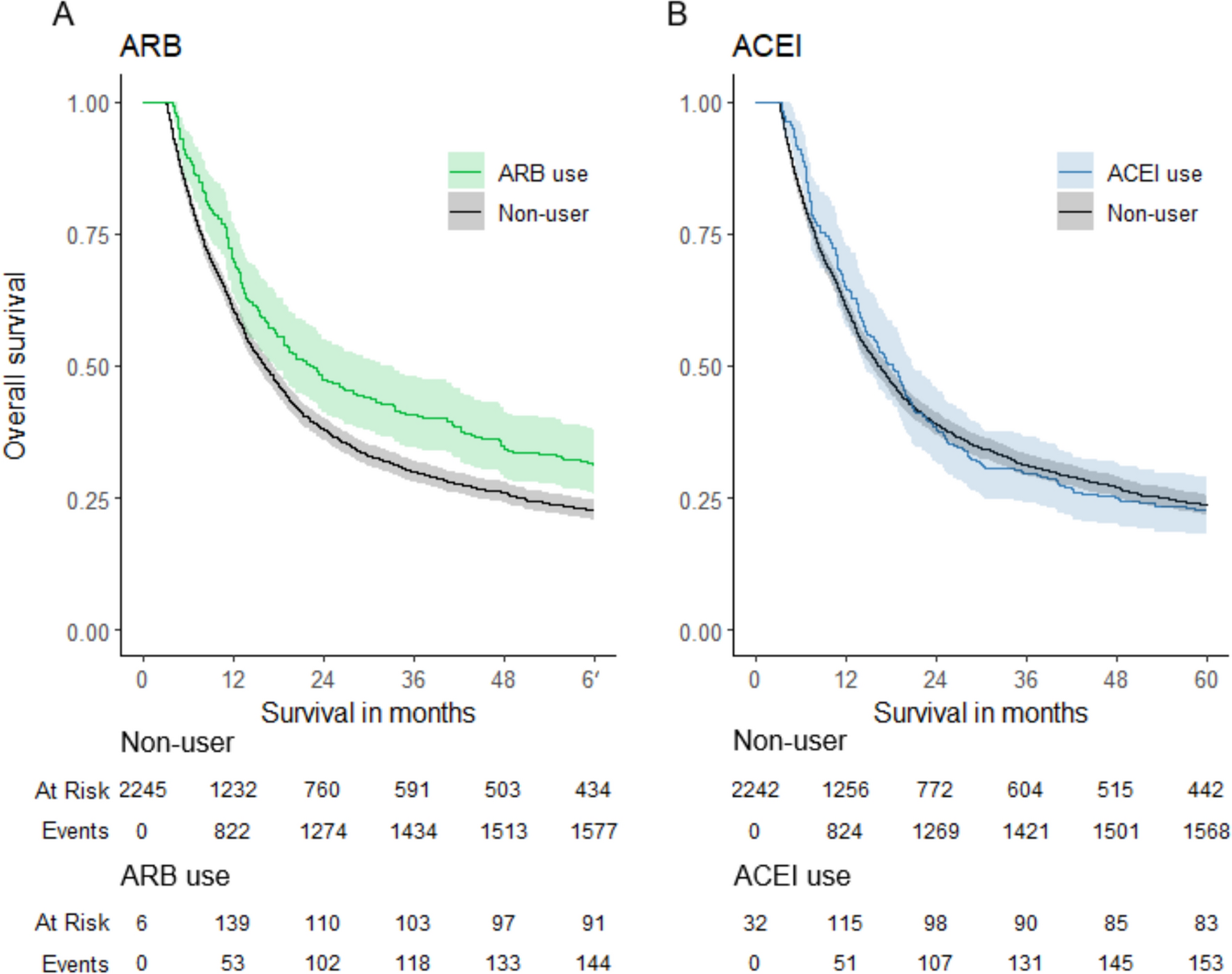
The findings of this study suggest that YAP activation plays a key role in the survival of DTPs during molecular-targeted therapy in GIST. In imatinib-sensitive GIST cells, DTPs induced by imatinib treatment exhibited increased nuclear localization and activity of YAP. YAP inhibitors demonstrated significant efficacy in targeting these cells. Moreover, the combination of imatinib and verteporfin sustained tumor growth suppression for up to 3 weeks following drug withdrawal, highlighting the potential utility of this combined approach in overcoming DTPs. Clinical data further supported these findings, showing an increased proportion of YAP-positive nuclei in preoperatively treated samples that responded well to imatinib, consistent with observations from cell-based experiments. These results address a persistent therapeutic challenge in GIST, where complete tumor eradication remains elusive despite advances in molecular-targeted therapies.
The relationship between DTPs and YAP has been reported in several preclinical and clinical studies of NSCLC [14, 15], suggesting that YAP activation following molecular-targeted therapy may represent a conserved phenomenon across diverse cancer types. In NSCLC-derived DTPs with EGFR or ALK mutations, targeted inhibitors have been shown to activate FAK signaling, which promotes nuclear localization of YAP [15]. In our previous study, we observed elevated FAK expression in GIST DTPs induced by imatinib [11]. These findings collectively provide indirect evidence that FAK may also be involved in the nuclear translocation of YAP in GIST DTPs. However, the present study did not directly investigate the mechanistic link between FAK and YAP activation, leaving it uncertain whether FAK signaling plays a causal role in YAP activation in this context. Considering the established role of FAK in NSCLC, it is logical to propose that FAK may similarly contribute to YAP-mediated resistance in GIST. Future studies are needed to validate this hypothesis and elucidate the underlying mechanisms.
Inhibition of KIT by imatinib has been shown to upregulate the pro-apoptotic factor BIM through transcriptional and post-translational mechanisms in GIST cell lines [23]. Conversely, DTPs induced by EGFR/MEK inhibition in EGFR-mutant NSCLC evade apoptosis by suppressing the expression of the pro-apoptotic factor BMF via the YAP/TEAD/SLUG complex [14]. Both BIM and BMF are involved in mitochondrial apoptosis, suggesting that YAP activation in DTPs may interfere with imatinib-induced cell death by modulating intrinsic apoptotic pathways [24]. Consistent with these findings, this study demonstrated that DTPs under imatinib treatment avoided apoptosis, whereas verteporfin effectively induced apoptosis in these cells. Notably, YAP inhibitors were ineffective in parental GIST cells, likely because cells primarily relied on the KIT signaling pathway. In contrast, DTPs exhibited a dependency on YAP for apoptosis evasion, underscoring the therapeutic potential of YAP inhibitors in this context.
In this study, two YAP inhibitors—verteporfin and XAV-939—were used, and both agents suppressed YAP activity in DTPs. However, the differences in pYAP expression observed in Fig. 3a likely stem from their distinct mechanisms of action. Verteporfin directly inhibits YAP/TAZ and YAP/TEAD interactions [25, 26], while XAV-939 inhibits tankyrase, stabilizing AMOT family proteins [18]. AMOT family proteins have been shown to activate LATS kinase, leading to YAP phosphorylation [27]. This mechanistic difference may account for the observed increase in pYAP expression in DTPs treated with XAV-939. Simultaneous inhibition of KIT and YAP in GIST represents a promising strategy to induce apoptosis in DTPs, potentially leading to complete tumor eradication.
The combination of imatinib and YAP inhibitors demonstrated significant efficacy in vitro and vivo. While imatinib monotherapy resulted in tumor regrowth after treatment cessation, verteporfin addition significantly delayed regrowth. IHC revealed an increased proportion of YAP-positive nuclei in tumors immediately after imatinib monotherapy, whereas combination therapy reduced this proportion. These findings confirm the establishment of DTPs in the animal model and highlight verteporfin’s inhibitory effects on YAP. However, the reduction in tumor volume observed in vivo was modest compared to the dramatic decrease in cell numbers seen in vitro, suggesting that imatinib’s in vivo efficacy is limited. This limitation may explain the eventual tumor regrowth observed in the combination therapy group. In this study, imatinib was administered for 12 d and verteporfin for only 3 d to align with in vitro protocols. Clinically, preoperative imatinib is typically administered for over 3 months, during which viable cells are rarely observed in well-responding cases. Extending the duration of drug administration in future animal studies could potentially yield different outcomes. Additionally, optimization of verteporfin dosing regimens is warranted.
In the verteporfin-only group, no significant weight loss was observed, while weight loss in the combination group was attributed primarily to imatinib, with no additional effects from verteporfin. This indicates that verteporfin can be safely administered in mice. Verteporfin, though primarily known as a YAP inhibitor, is clinically approved as a photosensitizer for photodynamic therapy in age-related macular degeneration [28]. While patients had to avoid direct sunlight for 48 h post-treatment, with eye and skin protection recommended, no serious adverse events were reported in randomized trials for age-related macular degeneration treatment [29, 30]. Its established safety profile and clinical use render verteporfin an attractive candidate for early adoption in cancer therapy, provided its antitumor effects are further validated.
This study has some limitations. First, our experiments were conducted using only one GIST cell line due to the unavailability of other imatinib-sensitive lines in Japan. Future studies utilizing additional GIST cell lines, including patient-derived samples, are needed to confirm these findings. Second, the limitation of this study is the lack of functional validation through YAP knockdown or knockout experiments. Although such approaches would provide strong support for the results obtained with YAP inhibitors, they were technically challenging to perform in DTPs. This is because DTPs represent a rare and transient cell population that emerges only under continuous exposure to molecular-targeted therapy, such as imatinib, making efficient gene delivery and selection difficult. The limited cell numbers, slow proliferation, and the need to maintain drug pressure during the process further complicate genetic manipulation. Future studies employing alternative strategies—such as establishing inducible gene silencing systems prior to DTP induction—may help overcome these technical challenges and enable direct functional assessment of YAP depletion in DTPs. Third, defining DTPs in clinical specimens poses challenges. Residual cells in cases responding to preoperative imatinib were classified as DTPs, but the possibility of including imatinib-resistant cells cannot be excluded, potentially confounding the results. Examining KIT mutation status pre- and post-treatment may aid in more accurate identification of DTPs. Lastly, while this study demonstrated the effectiveness of combination therapy with imatinib and verteporfin, the optimal YAP inhibitor for GIST treatment remains to be determined. Although YAP-dependent growth has been observed in malignant mesothelioma and solid tumors with neurofibromatosis 2 mutations, the American Association for Cancer Research recently reported on the efficacy of the oral YAP/TEAD inhibitor VT3989 in a phase 1 trial. Exploring other YAP inhibitors like VT3989 could contribute to optimizing combination therapies in GIST treatment.
This study demonstrated that YAP activity is elevated in DTPs following imatinib treatment in imatinib-sensitive GIST. Our findings suggest that combining imatinib with the YAP inhibitors may effectively target DTPs and reduce recurrence rates. By addressing the survival mechanisms of DTPs, this study provides a foundation for developing combination therapies to overcome resistance and facilitate the discontinuation of TKI therapy. These findings contribute to advancing curative treatment strategies for GIST and highlight the need for continued investigation into the molecular pathways underpinning drug tolerance.



Comments (0)