Study population
AVS patients were selected based on a retrospective analysis of clinical data (N = 80). Only patients with preoperative echocardiographic confirmation of severe AVS, irrespective of the number of cusps (two or three) and undergoing surgical valve replacement, were included. A severe AVS was defined as (1) a mean transvalvular pressure gradient > 40 mmHg; (2) an indexed aortic valve area < 0.6 cm2/m2; (3) echocardiographic evidence of relevant leaflet calcification (confirmed in the surgery report) in combination with left ventricle hypertrophy (indexed left ventricle mass ≥ 95 g/m2 for women and ≥ 115 g/m2 for men) not explained by hypertrophic cardiomyopathies or other conditions. The exclusion criteria included rheumatic AVS, severe aortic valve regurgitation, and lack of diagnostic echocardiography. The protocol was approved by the ethics committee of Centro Hospitalar Universitário de São João (reference CEC109-2020, 20/05/2020). All participants provided written informed consent. The study protocol abided by the principles outlined in the 1964 Declaration of Helsinki and its later amendments.
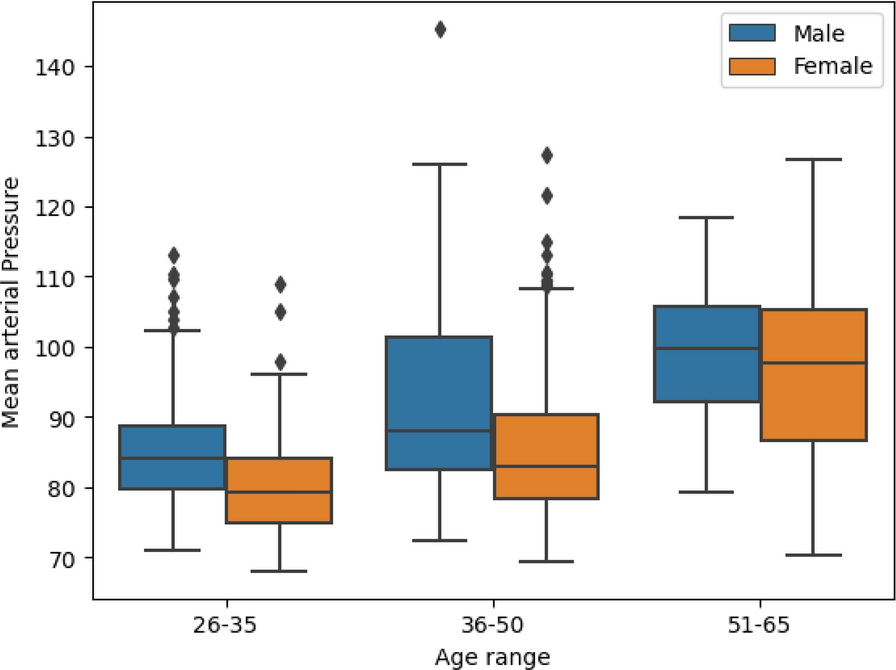
Clinical and demographic variables, including sex, age, BMI, comorbidities (e.g., hypertension, diabetes mellitus), and medication use, were obtained from medical records. Disease severity was assessed by transthoracic Doppler echocardiography. The mean (taoGmean) and maximal (taoGmax) aortic transvalvular pressure gradients and aortic valve area (AVA) were derived from the modified Bernoulli equation and the standard continuity equation, respectively. Whenever possible, the Doppler velocity index (DVI) was recorded. As an additional measure of disease severity, the left ventricle mass (LVM) was estimated following derivation of left ventricle end-diastolic dimension (LVEDD), posterior wall thickness (PWT), and interventricular septal thickness (IVST) from 2D echocardiograms during diastole with application of Deveraux’s formula (LVM = 0.8 × 1.04 × [(LVEDD + PWT + IVST)3– LVEDD3] + 0.6 g). Correct orientation of imaging planes, cardiac chambers dimension, and function measurements were performed according to the European Association of Echocardiography/American Society of Echocardiography recommendations [21].
Sample collection and processing
Aortic valves were obtained as a by-product of surgical AVR, isolated or concomitant with other procedures, in most cases, coronary artery bypass grafting, without any additional risk to the patients. Each sample was immediately immersed in cardioplegic solution (Custodiol®) and kept at 4 °C until delivery to the laboratory (< 2 h). Each valve leaflet was washed thrice with phosphate-buffered saline (PBS) to remove residual blood. Samples were weighed to obtain an indirect measure of calcium content. A representative transversal cut was obtained for histology. The remaining material was grossly fragmented and randomly processed for RNA or proteomics/protein analyses. For the former, the fragments were preserved in RNA later™ (Sigma) and incubated overnight at 4 °C before storage at −80 °C. For the latter, the fragments were directly stored at −80 °C until further processing.
Histology
Aortic valve tissue was automatically processed using a LEICA HistoCore Pearl processor. AV tissue was fixed by diffusion of 4% (V/V) buffered formaldehyde, followed by dehydration with ethanol in crescent concentrations, clearing with xylene, and paraffin impregnation. Paraffin-embedded samples were then included in blocks in the LEICA HistoCore Arcadia. After refrigeration, the paraffin blocks were cut into 3 μm-thick sections using a microtome (LEICA RM2125 RTS). The top sections were discarded, and when the sample emerged, it was subjected to gross decalcification by incubation in a decalcifying solution with ethylenediamine tetraacetic acid (Osteosens, Biognost®, Croatia) for 5 min. Then, sample sections were cut and placed on sequential water baths, first at room temperature (RT) and then at 40 °C, to unfold the tissue slices. Finally, the sections were allowed to dry on glass slides.
Before staining, slides were deparaffinised and rehydrated. First, tissue paraffin sections were heated at 60 °C for 30 min to dissolve paraffin. The slides were then deparaffinised twice with fresh xylene (10 min + 5 min) and dehydrated with decreasing ethanol concentrations (100%, 80%, and 70%, 2 min each). Finally, the slides were rinsed with tap water for 5 min for hydration. All staining procedures were performed at RT.
Slides were stained with Haematoxylin and Eosin (H&E) to evaluate the general morphology and tissue architecture. First, slides were placed in modified Harris haematoxylin (Biognost®) for 8 min and then washed with tap water. The slides were briefly immersed in acid alcohol (hydrochloric acid 37% in 70% ethanol, 1:100) and washed with tap water between the stains. Next, the slides were placed in alcoholic eosin Y 1% (0.2–0.5% acetic acid, 70–90% ethanol, Biognost®) for 2 min. Finally, the preparations were dehydrated with ethanol (90% for 30 s and 99.5% for 1 min) and cleared with xylene for 5 min.
To assess collagen deposition in the aortic valve, the slides were stained with Red Sirius 0.1% (Direct Red 80, in 1.3% picric acid solution, Sigma) for 90 min and then briefly rinsed in acid water (0.5% glacial acetic acid). Next, the slides were dehydrated in three ethanol solutions of 99.5% and deparaffinised twice with xylene for 5 min.
To evaluate the extent of calcification, the slides were placed in 2% Alizarin Red S solution (Sigma) for 5 min and briefly rinsed in water. Next, the slides were sequentially treated with acetone for 1 min and acetone: xylene (1:1) for 1 min. Finally, the slides were cleared twice with xylene for 5 min.
For visualisation and image acquisition, the slides were mounted with Entellan® and observed under an optical microscope (Zeiss Axio Scope.A1, Germany) equipped with a photographic camera (Olympus XC30, Tokyo, Japan). For H&E staining, photographs were taken under 25× magnification, while for Red Sirius and Alizarin Red S staining, a 40× magnification was used. Fibrosis and calcific load were analysed using Image Pro Plus software by an observer and then reviewed by an expert. To minimise potential biases and ensure a standardised and impartial assessment of fibrosis and calcification, the observer and the expert were blind to the patient’s sex until all sections were analysed. This analysis involved calculating the tissue area in each region-of-interest, followed by assessment of the stained areas, divided by the tissue area. Eight regions-of-interest were evaluated for each patient, and the results were averaged. Fibrotic and calcific areas were expressed relative to the total tissue area (%).
ProteomicsProtein digestion and sample clean-up
Fifty AV samples (25 men) participants were selected for proteomic analysis, ensuring matching based on key clinical covariates [22]. Fifty µg of protein of each sample was precipitated with 6 volumes of cold acetone, and the pellet was dissolved in 50 µL of 6 M urea and 200 mM ammonium bicarbonate. Then, samples were reduced with dithiotreitol (150 nmol, 37 °C, 60 min) and alkylated in the dark with iodoacetamide (300 nmol, 25 °C, 30 min). The resulting protein extract was first diluted to 2 M urea with 200 mM ammonium bicarbonate for digestion with endoproteinase LysC (1:100 w/w, 37 °C, over 6 h, Wako, cat #129–02541) and then diluted 2-fold with 200 mM ammonium bicarbonate for trypsin digestion (1:100 w/w, 37 °C, over 6 h, Promega, cat #V5113). After digestion, the peptide mix was acidified with formic acid and desalted with a MicroSpin C18 column (The Nest Group, Inc.) before LC-MS/MS analysis.
Tissue homogenisation and protein extraction
Fifty aortic valves were homogenised following a standard operating procedure we previously optimised for the proteomic characterisation of calcified valves [22]. Briefly, the tissue was disrupted with zirconium oxide beads (2.8 mm, Precellys®, Bertin Technologies) using a bead-beating system (Minilys, Bertin Instruments). Approximately 30 mg of tissue was combined with ~ 1.2 g of beads in O-ring tubes. Ice-cold RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, Thermo Fisher), supplemented with 1 mM EDTA, a protease inhibitor cocktail (Halt PIC, Thermo Fisher), and a phosphatase inhibitor cocktail (PhosSTOP, Roche) was added at a 10 µL/mg ratio. Then, the samples were homogenised twice, at maximum speed (5,000 rpm) for 30 s, in the Minilys instrument, with a 5-min cool down on ice between cycles. The sample was centrifuged at 12,000 rpm (13,680 × g) for 15 min at 4 °C to obtain a clear supernatant. The sample was aliquoted in 100µL fractions and stored at −80 °C until further analysis.
Protein concentration was estimated using the detergent-compatible (DC) method (Bio-Rad Laboratories), following the manufacturer’s instructions. Bovine serum albumin was used as a standard. All samples and standards were prepared in duplicate, and quadruplicate absorbance readings were averaged to obtain the concentration of each sample.
LC-MS/MS analysis
Samples were analysed using an Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific) coupled to an EASY-nLC 1200 (Thermo Fisher Scientific). Peptides were loaded directly onto the analytical column and separated by reversed-phase chromatography using a 50-cm column with an inner diameter of 75 μm, packed with 2 μm C18 particles (Thermo Fisher Scientific, cat #ES903).
Chromatographic gradients started at 95% buffer A (0.1% formic acid in water) and 5% buffer B (0.1% formic acid in 80% acetonitrile) with a flow rate of 300 nL/min and gradually increased to 25% buffer B and 75% A in 105 min and then to 40% buffer B and 60% A in 15 min. After each analysis, the column was washed for 10 min with 100% buffer B.
The mass spectrometer was operated in positive ionisation mode with nanospray voltage set at 2.4 kV and source temperature at 305 °C. The instrument was operated in data-independent acquisition (DIA) mode, with full MS scans over a mass range of m/z 500–900 with detection in the Orbitrap at a resolution of 120,000. The auto gain control (AGC) was set to 1e6, and a maximum injection time of 246 ms was used. In each cycle of DIA analysis, following each survey scan, 40 consecutive windows of 10 Da each were used to isolate and fragment all precursor ions from 500 to 900 m/z. A normalised collision energy of 28% was used for higher-energy collisional dissociation (HCD) fragmentation. MS2 scan range was set from 350 to 1850 m/z with detection in the Orbitrap at a resolution of 30,000. The AGC was set to 1e6, and a maximum injection time of 54 ms was used.
Digested bovine serum albumin (New England Biolabs, cat #P8108S) was analysed between each sample to avoid sample carryover and ensure the instrument’s stability. Qcloud was used to control instrument longitudinal performance during the project [23].
Protein identification and quantification
Acquired spectra were analysed using a library-free strategy with DIA-NN (v.1.7.12) [24]. A Swiss-Prot human database (January 2021, 20395 entries) plus a list of common contaminants [25] was used as a reference proteome. For peptide identification, trypsin was chosen as the enzyme and up to one missed cleavage was allowed. Methionine oxidation was used as variable modification, whereas carbamidomethylation of cysteine was set as fixed modification. A false discovery rate (FDR) was set to a maximum of 1% in peptides. Precursor and fragment ion m/z mass range were adjusted to 500–900 and 350–1850, respectively. Default settings were used for the other parameters (the full search report is provided as part of Supplementary File 1). Proteins were deemed identified when passing a 1% FDR threshold and those that, in addition, were quantified in at least 75% of the samples of one sex were regarded as confidently quantified. Given the approach used (DIA), no further filter was applied to the number of peptides sequenced per protein.
The raw proteomics data have been deposited to the PRIDE [26] repository with the dataset identifier PXD051201.
BioinformaticsFunctional enrichment analysis
The clusterProfiler package was used to map gene ontology terms pertaining to Biological Processes (BP), Molecular Functions (MF), and Cellular Components (CC) to the DEPs. The human library was obtained on 16 March 2022, and only terms surpassing a 5% FDR were considered.
In addition, cell-type enrichment analysis of the DEPs was conducted using the Enrichr webtool (https://maayanlab.cloud/Enrichr/) to ascertain which cell types were mainly enriched in the valves of men and women. Proteins were mapped to the Human Gene Atlas database, and only cell types with an adjusted p-value < 0.05 were considered.
Protein-Protein interaction analysis
To identify the most influential proteins in the definition of sexual dimorphism in AVS, a protein-protein interaction (PPI) analysis was performed using the STRING application (v.11.5), available for Cytoscape (v.3.9.1). Only the DEPs were considered for this analysis, and all known interactions with a medium confidence score of 0.4 (default) were retrieved. Topological network analysis was run using Cytoscape built-in tools to calculate node degree and betweenness centrality and identify protein hubs and bottlenecks.
Identification of proteins differentially expressed between sexes
Protein data were analysed in R (version 4.2.3) with the package DEP (Differential Enrichment Analysis of Proteomics Data) to identify differentially expressed proteins. A variance-stabilising normalisation method was applied to the protein quantification raw data [27]. Given that the DIA method results in fewer missing values, no data imputation was performed. Missing values were random (no sex-defined cluster was observed) and corresponded to proteins close to the detection limit (Supplementary Figs. 1 and 2). A differential enrichment test based on protein-wise linear models and empirical Bayes statistics using limma was applied to prioritise the most significantly changed proteins, considering an adjusted p-value of ≤ 0.2. Only unambiguously identified proteins were considered as DEPs. Protein groups assigned to more than one UniProt ID were removed. The standardised mean difference (Cohen’s d) was used as an additional criterion to select the most relevant proteins explaining sexual dimorphism. DEPs were considered only when they showed a large effect size (Cohen’s d > 0.8).
RNA extraction and qRT-PCR
RNA was extracted using the Aurum™ Total RNA Fatty and Fibrous Tissue Kit (catalogue #732–6830, Bio-Rad), following the manufacturer’s instructions. This kit was chosen due to its superior performance concerning the extraction yield and RNA quality in calcified valve tissue [28].
RNA was extracted from approximately 100 mg of tissue using 2.8 mm zirconium oxide beads in the same instrument used for protein extraction (Minilys). 1 mL of PureZOL was added to each sample, tissue disruption was performed at maximum speed (5,000 rpm) in 3 cycles of 30 s, and the sample was incubated on ice between cycles. The collected lysate was incubated at RT for 5 min and centrifuged at 12,000 × g for 10 min at 4 °C to remove insoluble material. The supernatant was transferred to a new microtube and vigorously mixed with 0.2 mL of chloroform for 15 s. The samples were incubated for 5 min at RT, periodically mixed, and centrifuged at 12,000 × g for 15 min at 4 °C to separate the phases. The RNA contained in the aqueous phase was immediately transferred to a new tube and thoroughly mixed with 400 µL 70% ethanol by pipetting. The samples were transferred to an RNA-binding mini-column and centrifuged at 12,000 × g for 1 min at RT to purify the RNA. A low-stringency wash solution (700 µL) was added to the RNA-binding mini-column, and the samples were centrifuged again for 30 s at RT. Each column was incubated with 80 µL of diluted DNase I for 15 min to remove contaminating genomic DNA. Next, 700 µL of high-stringency wash solution was added to the RNA-binding mini-column, and centrifugation was performed at 12,000 × g for 30 s at RT. A final wash with 700 µL of low-stringency solution was completed, and the samples were centrifuged twice at 12,000 × g for 2 min at RT. To elute RNA, the mini-columns were incubated twice with 25 µL of elution solution for 1 min and centrifuged at 12,000 × g for 2 min at RT. The purified RNA was stored at −20 °C until further use.
RNA concentration and purity were measured using the optical density at 260/280 nm and 260/230 nm in a NanoDrop spectrophotometer (Thermo Scientific). RNA integrity was verified using electrophoresis.
From an initial set of 50 samples, RNA was successfully extracted from 47 (24 men and 23 women). Eleven dysregulated proteins were selected for validation at the transcript level, including AIF1, ANPEP, CD163, CD74, DNAJA1, F13A1, GPX1, NOX2 (or CYBB), OSBP1, PFKL, and STEAP4, in addition to a panel of 7 genes (COL1A1, COL3A1, MMP2, MMP9, TGFB, TIMP1, and TIMP2) associated with fibrosis regulation.
After RNA extraction, cDNA (100 ng/µL) was synthesised using a SensiFAST™ cDNA Synthesis Kit (Bioline). A mastermix of the RNA sample, 5x TransAmp buffer, reverse transcriptase enzyme, and DNase/RNase free water was prepared and transferred to the Bio-Rad T100™ Thermal Cycler. Reactions were controlled using the following protocol: 10 min at 25 °C for primer annealing, 15 min at 42 °C for reverse transcription, and 5 min at 85 °C to inactivate reverse transcriptase.
Quantitative polymerase chain reaction (qPCR) reactions were run in duplicate in a 10 µL reaction volume, including 1 µL of cDNA, 5 µL 2×SensiFAST™SYBR Hi-ROX Mix (Bioline), and 0.4 µL of primers (Supplementary Table 1). The RT-qPCR reactions were monitored with the PikoReal™ 96 and QuantStudio™ 5, Real-Time PCR Systems (Thermo Scientific™), using the following protocol: 3 min at 95 °C for polymerase activation, followed by 40 cycles of 15 s at 95 °C for denaturation, 30 s at 60 °C for annealing, and 30 s at 72 °C for chain extension. Melting curve analysis was performed from 65 to 95 °C in 0.5 °C increments.
Before quantification of gene expression, the PCR efficiency of each gene, including the internal control gene (18 S RNA), was determined, and it was confirmed that they were identical.
The target gene expression was normalised to 18 S rRNA gene expression. Gene expression was analysed using a comparative method.
Immunohistochemistry
The differential expression of CD74 and CD163, markers of immune cells, in AV tissue, was validated by immunohistochemistry in 12 patients (6 women). Tissue sections were placed in an antigen retrieval solution (0.1 M citrate buffer, composed of 0.0825 M sodium citrate dihydrate and 0.0175 M citric acid, pH 6.0) and heated in a microwave for 20 min. The slides were allowed to cool to RT, and the tissue sections were outlined with a hydrophobic pen (IHC PAP pen, Enzo). The sections were then incubated in 0.3% H2O2 (UltraVision hydrogen peroxide block, Thermo Scientific) for 10 min to quench the endogenous peroxidase activity. The slides were washed with PBS, incubated for 5 min with UltraVision protein blocking solution (Thermo Scientific), and washed again with PBS. Next, the tissue sections were incubated with the respective unlabelled primary antibodies, mouse monoclonal anti-CD74 (1:250 in PBS, sc-6262, Santa Cruz Biotechnology) and mouse monoclonal anti-CD163 (1:200 in PBS, sc-20066, Santa Cruz Biotechnology) for 30 min at RT, and washed with PBS. The slides were then incubated with a biotinylated secondary antibody (UltraVision Primary Antibody Amplifier Quanto, Thermo Scientific) for 10 min at RT and washed with PBS. To enhance the signal, sections were incubated with streptavidin-horseradish peroxidase conjugate (UltraVision HRP Polymer Quanto, Thermo Scientific) for 15 min at RT. The slides were washed thoroughly with deionised water and incubated with the corresponding substrate solution (3,3′-Diaminobenzidine, DAB, Thermo Scientific) for 5 min. Haematoxylin (Thermo Scientific) was used as the counterstain. Finally, the sections were dehydrated with ethanol, cleared with xylol, and mounted using Entellan®. The primary antibody was replaced with PBS as a negative control.
Slides were photographed under the same microscope under 200× magnification. The protein signal was analysed using the Image Pro Plus software and expressed as the number of DAB spots relative to the total tissue area.
ELISA
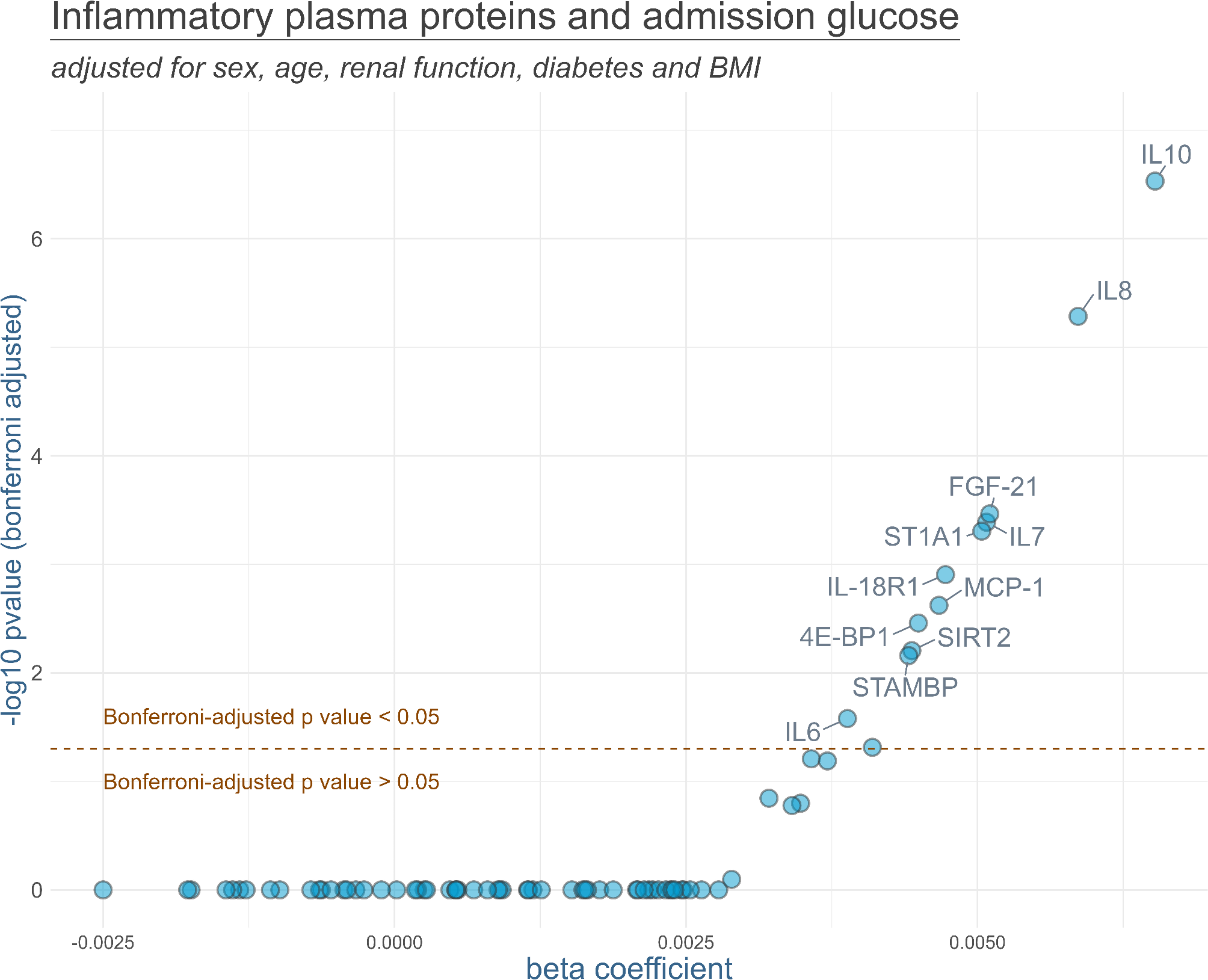
Differences in the protein levels of GPX1 (ref. A78193) and NOX2 (ref. A78533) between sexes were further confirmed by enzyme-linked immunosorbent assay (ELISA), following the manufacturer’s instructions (Antibodies.com, United Kingdom). AV lysates prepared for proteomics and 30 extra samples (N = 80) were diluted 10× and 2× with sample dilution buffer (provided by the kit) for GPX1 and NOX2 quantification, respectively. The absorbance of the standards and samples was recorded at a wavelength of 450 nm. Protein concentration was interpolated from the best-fitting curve (sigmoidal in both cases).
Statistical analysis
Clinical and demographic data was analysed in R (version 4.3.1), and the differences between men and women were queried with the package gtsummary (version 1.7.2). Continuous variables are presented as medians (interquartile ranges), and categorical data as absolute and relative frequencies. The proteome was also analysed in R (see above). To analyse the histological and specific molecular data, we used GraphPad Prism 9.5.1. Differences between sexes in fibrosis, calcification, and immunohistochemistry staining were inspected by an unpaired Mann-Whitney U test. Gene expression data (qRT-PCR) were normalised to RNA18SN1, and protein quantification data (ELISA) were normalised to total protein. Outliers in the PCR and ELISA data were removed using the ROUT method, with the highest level of stringency (Q = 0.1%). Differences in gene expression and protein levels were investigated using unpaired t-tests. Results were considered significant at p < 0.05.





Comments (0)