Preparation of FFPE tissue sectionsFFPE tissue sectioning and review
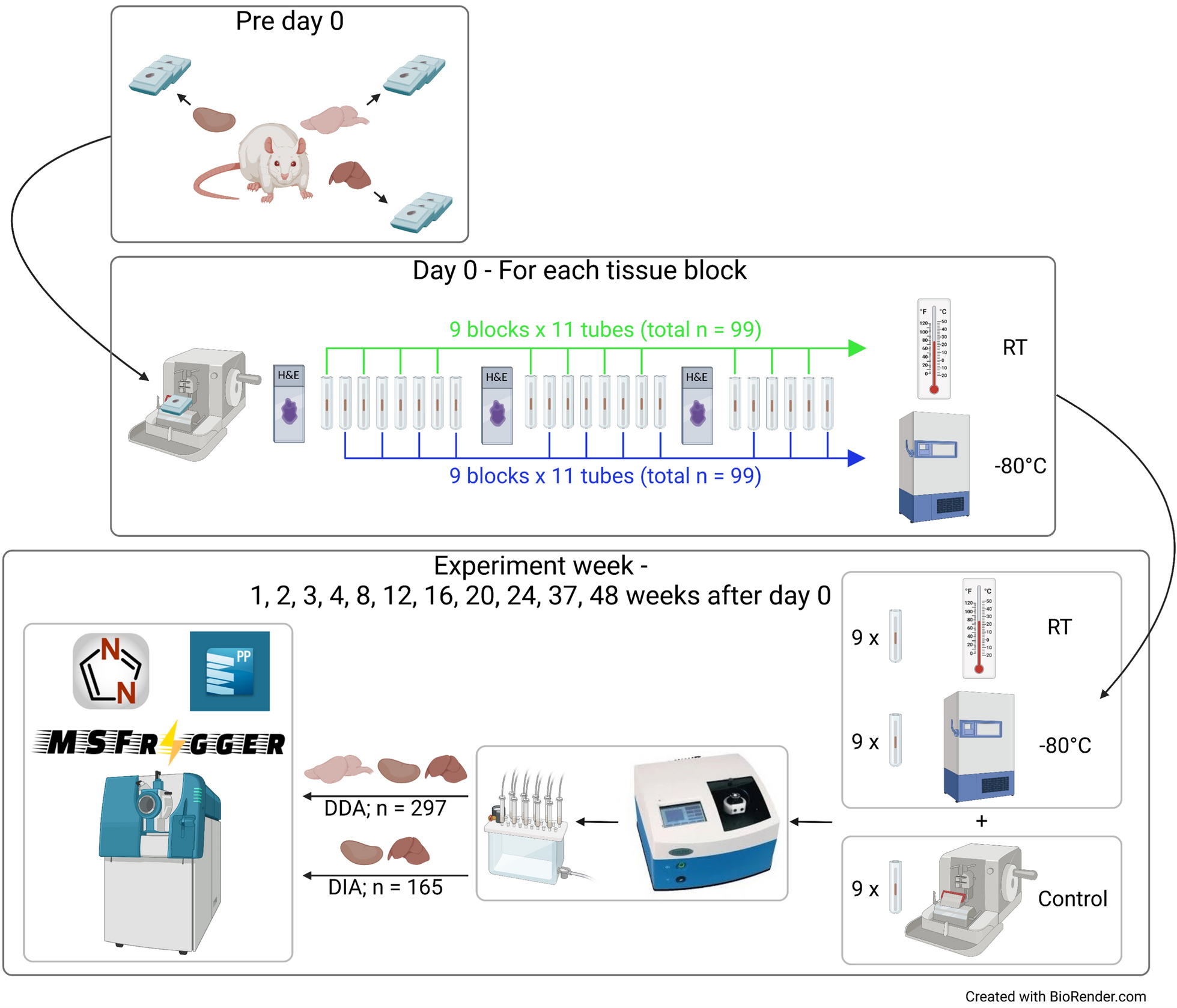
Brain, kidney and liver were harvested from an adult male Wistar rat. Tissues were fixed in 10% (v/v) neutral buffered formalin for 24 h and then transferred to 70% (v/v) ethanol prior to being embedded into paraffin blocks. Use of experimental animal tissues was approved by the Animal Care and Ethics Committee for Children’s Medical Research Institute/Sydney Children’s Hospital Network, Sydney, Australia, approval number C116.
To ensure that there would be sufficient comparable tissues for the entire experiment, three blocks were prepared from each tissue (total n = 9). A section from each tissue block was prepared for each timepoint and storage temperature so that each tissue was represented in triplicate. Tissue blocks were sectioned using a rotary microtome (Leica Biosystems, Nussloch, Germany) to collect a 4 µm section onto a glass slide for haematoxylin and eosin (H&E) staining, followed by 10 µm sections for proteomic analysis. Each 10 µm section was placed into a Barocycler MicroTube (Pressure Biosciences Inc., MA, USA) and then stored in an enclosed tube. Sections were numbered sequentially from the surface of the block. H&E stained slides were scanned to digital images (Zeiss Axio Scan Z1, Oberkochen, Germany) and viewed with Zen 3.1 (Zen lite) viewing software (Zeiss). Tissue size was measured using a manual outline trace and automated area measurement (in mm2).
Experiment schedule
On day 0, 22 sections were cut from each of the nine tissue blocks for proteomic analysis (total n = 198). Odd numbered samples from each block were stored at RT (around 22°C) and even numbered samples were stored at − 80°C. An H&E slide was prepared from each block surface after every eight 10 µm sections to track changes in tissue area. Tissue blocks were stored at RT throughout the experiment. Eleven experiment days were scheduled on 1, 2, 3, 4, 8, 12, 16, 20, 24, 37 and 48 weeks after day 0. On each experiment day, an H&E slide and a 10 µm section were prepared from each of the nine tissue blocks. These samples were combined with two sets of nine tubes that were stored at RT and − 80°C from day 0 to form a sample set (n = 27).
Oropharyngeal tissue sections
Human oropharyngeal tissue sections (10 µm) were prepared at the Princess Alexandra Hospital (PAH, Brisbane, QLD, Australia) with ethics approval obtained through the Metro South Human Research Ethics Committee at PAH (HREC/14/QPAH/54).
LC–MS analysis of tissue sectionsSample preparation
All samples were processed for LC–MS using the Heat ‘n Beat (HnB) method [18]. Paraffin wax was removed from tissue samples with heptane-methanol for 10 min at 30°C then samples were dried for 10 min and suspended in 5% (w/v) sodium deoxycholate (SDC) in 100 mM triethylammonium bicarbonate (TEAB), 4.2 mM tris(2-carboxyethyl)phosphine (TCEP) and 16.75 mM iodoacetamide (IOA) for 7 min at 95°C. Zirconium beads (Benchmark Scientific, NJ, USA), 50 µL of Rapid Digest Buffer (Promega, Alexandria, NSW, Australia), 1 µg of Rapid Trypsin/Lys-C (Promega) and 1 unit of Benzonase nuclease (Sigma-Aldrich, Castle Hill, NSW, Australia) were added prior to 1 min homogenisation in a Beadbug homogeniser (Benchmark Scientific, NJ, USA) at 3,800 rpm. Further lysis and digestion were carried out in a Barocycler 2320EXT (Pressure BioSciences Inc.) for 30 min at 56°C. The digests were acidified with 5 µL of formic acid (FA) to precipitate the SDC before being centrifuged for 15 min (18,000 × g, 4°C). Peptides were extracted from the supernatant using Oasis PRiME HLB 1 cc (30 mg sorbent) SPE cartridges (Waters, Rydalmere, NSW, Australia) and their concentration was determined using A280 nm with an Implen nanophotometer NP80 (Implen, München, Germany).
DDA MS acquisition
All samples (n = 297) were analysed in DDA mode on multiple mass spectrometers over 48 weeks. Samples from the same experiment day were analysed on the same mass spectrometer in randomised order. An Eksigent nanoLC 425 HPLC (SCIEX, MA, USA) operating in microflow mode, coupled online to a TripleTOF 6600 (SCIEX) was used for all analyses. Peptide digests (2 µg) were spiked with retention time standards (non-human internal spike peptides, Biognosys iRT peptides and PROCAL peptides) and injected onto a C18 trap column (SGE TRAPCOL C18 G203 300 µm × 100 mm) and desalted for 5 min at 8 µL/min with solvent A (0.1% [v/v] FA). The trap column was switched in-line with a reversed-phase capillary column (SGE C18 G203 300 µm × 250 mm, ID 3 µm, 200 Å), maintained at 40 °C. The flow rate was 5 µL/min. The gradient started at 2% solvent B (99.9% [v/v] acetonitrile, 0.1% [v/v] FA) and increased to 10% over 5 min. This was followed by an increase of solvent B to 25% over 60 min, then a further increase to 40% for 5 min. The column was washed with a 4 min linear gradient to 95% solvent B held for 5 min, followed by a 9 min column equilibration step with 98% solvent A. The LC eluent was analysed using the TripleTOF 6600 system equipped with a DuoSpray source and 50 µm internal diameter electrode and controlled by Analyst 1.7.1 software. The following parameters were used: 5500 V ion spray voltage; 25 nitrogen curtain gas; 100 °C TEM; 20 source gas 1; 20 source gas 2. The 90 min DDA acquisition, consisted of a survey scan of 200 ms (TOF–MS) in the range 350–1250 m/z to collect the MS1 spectra and the top 40 precursor ions with charge states from + 2 to + 5 were selected for subsequent fragmentation with an accumulation time of 50 ms per MS/MS experiment for a total cycle time of 2.3 s and MS/MS spectra were acquired in the range 100–2000 m/z.
DIA MS acquisition
Kidney (n = 66) and liver (n = 99) samples were analysed in DIA mode after all the samples were processed and analysed by DDA. For the DIA acquisition, peptide spectra were acquired with the LC–MS/MS method described above for DDA, using 100 variable windows. The parameters used were: lower m/z limit 350; upper m/z limit 1250; window overlap (Da) 1.0; CES was set at 5 for the smaller windows, then 8 for larger windows, and 10 for the largest windows. MS2 spectra were collected in the range 100–2000 m/z for 30 ms in high resolution mode and the resulting total cycle time was 3.2 s.
Data processing and analysisProteinPilot
DDA data were searched using ProteinPilot version 5.0 (SCIEX) with Paragon against a rat SWISSPROT database (downloaded on 17 July 2019) that included canonical sequences only. It contained 8,071 proteins and 16,142 peptides with manually inserted retention time standards. The following search parameters were used: sample type—identification; Cys alkylation—iodoacetamide; digestion—trypsin; instrument—TripleTOF 6600; search effort—thorough ID; detected protein threshold—0.05 (10.0%), the false discovery rate (FDR) analysis was selected and set at 1%.
MSFragger
Raw MS files were converted to mzML format using MSConvertGUI version 3.0 [30] with peak peaking vendor MS = level 1–2. MSFragger version 3.1.1 [31] was run on FragPipe version 14.0, with Philisopher version 3.3.12 and Python version 3.8.3. All DDA data were subjected to an open search using default open search settings with RAM set as 64, parallelism 60 and regular MS was selected. The rat SWISSPROT canonical database used for ProteinPilot search was also used for MSFragger search with decoys added. Fragment mass tolerance and precursor true tolerance were set as 50 ppm. Crystal-C, PeptideProphet, ProteinProphet and PTM-Shepherd were selected and run using defaults for open search. DDA runs from weeks 1, 12, 24, 37 and 48 were subjected to a closed search against the same rat database using default LFQ settings. For MSFragger analysis, the closed search default config was selected, fragment mass tolerance and precursor true tolerance were set as 20 ppm. A list of variable modifications that was identified from the open search which are common for FFPE samples was included in the search and a maximum of three variable modifications was allowed for each peptide (Table 1). PeptideProphet, ProteinProphet and MS1 Quantification were selected and run using defaults for closed search. A spectral library was generated from the search results using EasyPQP.
Table 1 Modifications commonly associated with FFPE samples and reported in the literature were identified using an MSFragger open search of DDA runs
DIA-NN
DIA data were searched using Data-Independent Acquisition by Neural Networks (DIA-NN) version 1.7.12 [32] against the spectral library generated from the closed search on MSFragger. The following settings were used: protease—Trypsin/P with 1 missed cleavage; maximum number of variable modifications—0; N-term M excision and C carbamidomethylation were selected; peptide length range 7–30; precursor m/z range 400–1250; fragment ion m/z range 100–2000; precursor FDR set at 1%; cross-run normalisation—RT-dependent; quantification strategy—robust LC (high precision). Fixed modifications were allowed for the search, including the nine FFPE-specific modifications identified from MSFragger open search (Table 1). LOESS normalisation was performed on the peptide matrix before further analysis.
Statistical analysis
For the statistical analysis of DDA data, paired t-test followed by Bonferonni-Dunn’s multiple comparisons was performed using GraphPad Prism version 9.4.0 for Windows (CA, USA). Paired t-test was also performed on DIA data using Python modules scipy.stats [33] and statsmodels.stats.multitest [34], with Bonferonni method being selected for p-value correction.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [35] partner repository with the dataset identifier PXD054596.





Comments (0)