
Remember me



Diabetes and osteoporosis are chronic diseases with high prevalence worldwide, seriously affecting patients’ quality of life and health prognosis [1, 2]. The risk of fractures is significantly increased in patients with diabetes, especially in patients with type 2 diabetes, who are at higher risk of reduced bone strength and bone quality despite having normal or slightly higher bone density [3, 4]. In recent years, more and more studies have shown that diabetes not only affects bone metabolism through hyperglycemia and insulin resistance, but also interferes with the bone remodeling process through mechanisms such as chronic low-grade inflammation and oxidative stress [5, 6]. These pathological mechanisms play a key role in diabetes-induced osteoporosis, promoting the activation of osteoclasts and impaired osteoblast function, thereby accelerating bone resorption and inhibiting bone formation [7].
Existing research on diabetic osteoporosis mainly focuses on metabolic changes in a hyperglycemic environment, such as the accumulation of advanced glycation end products (AGEs) and their direct damage to bone tissue [8, 9]. However, the mechanism of imbalance between inflammation and bone remodeling has not been fully studied. Chronic inflammation, especially the low-grade chronic inflammatory response in diabetes, continues to act on bone metabolism through inflammatory factors such as TNF-α and IL-6, further exacerbating the risk of osteoporosis [10,11,12]. In addition, oxidative stress in patients with diabetes is closely related to bone metabolism disorders and may play a synergistic role in the imbalance between inflammation and bone remodeling [13, 14]. In recent years, several clinical guidelines have also emphasized the need to evaluate bone health in diabetic patients. For example, the American Diabetes Association (ADA) and the International Osteoporosis Foundation (IOF) have identified type 2 diabetes as a clinical risk factor for fragility fractures, regardless of normal bone mineral density, and recommend adjusting fracture risk assessments accordingly [[3, 15]. Furthermore, the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO) and the American Association of Clinical Endocrinologists (AACE) support integrated strategies that simultaneously address both glucose and bone metabolism in affected patients [16, 17].Therefore, an in-depth understanding of the pathological mechanisms of the interaction between diabetes and osteoporosis is crucial to developing effective prevention and treatment programs. This review will systematically explore the interactive mechanism between diabetes and osteoporosis, focusing on the role of chronic inflammation and bone remodeling imbalance, evaluate existing treatments, and look forward to possible future research directions.
Pathophysiological mechanisms of diabetes and osteoporosisInsulin resistance and bone metabolism disordersInsulin is not only a key hormone in blood sugar regulation, but also regulates bone metabolism through complex signaling pathways [18]. Under normal circumstances, insulin maintains bone metabolic balance by activating the proliferation and differentiation of osteoblasts, inhibiting the activity of osteoclasts [19, 20]. However, patients with diabetes are often accompanied by insulin resistance, which causes bone cells to be significantly less sensitive to insulin, disrupting this balance [21, 22]. Osteoblasts are less responsive to insulin signaling, resulting in reduced bone formation, while osteoclast activity is increased, leading to increased bone resorption [23, 24]. This metabolic disorder directly weakens the quality and structure of bone tissue and increases the incidence of osteoporosis [25].
Direct damage to bone tissue caused by high blood sugarLong-term hyperglycemia in diabetic patients has a direct destructive effect on bones [26]. Hyperglycemia not only affects the function of osteoblasts and osteoclasts, but also changes the normal synthesis of bone matrix proteins, resulting in impaired bone mineralization [27, 28]. The quality of proteins such as collagen in the bone matrix decreases, weakening the overall strength of the bone tissue [29]. The oxidative stress that accompanies hyperglycemia exacerbates this condition, increasing bone resorption and accelerating bone loss [30]. In addition, the impact of a high-glycemic environment on surrounding soft tissues such as cartilage and ligaments further weakens bone function and increases the risk of fractures [31].
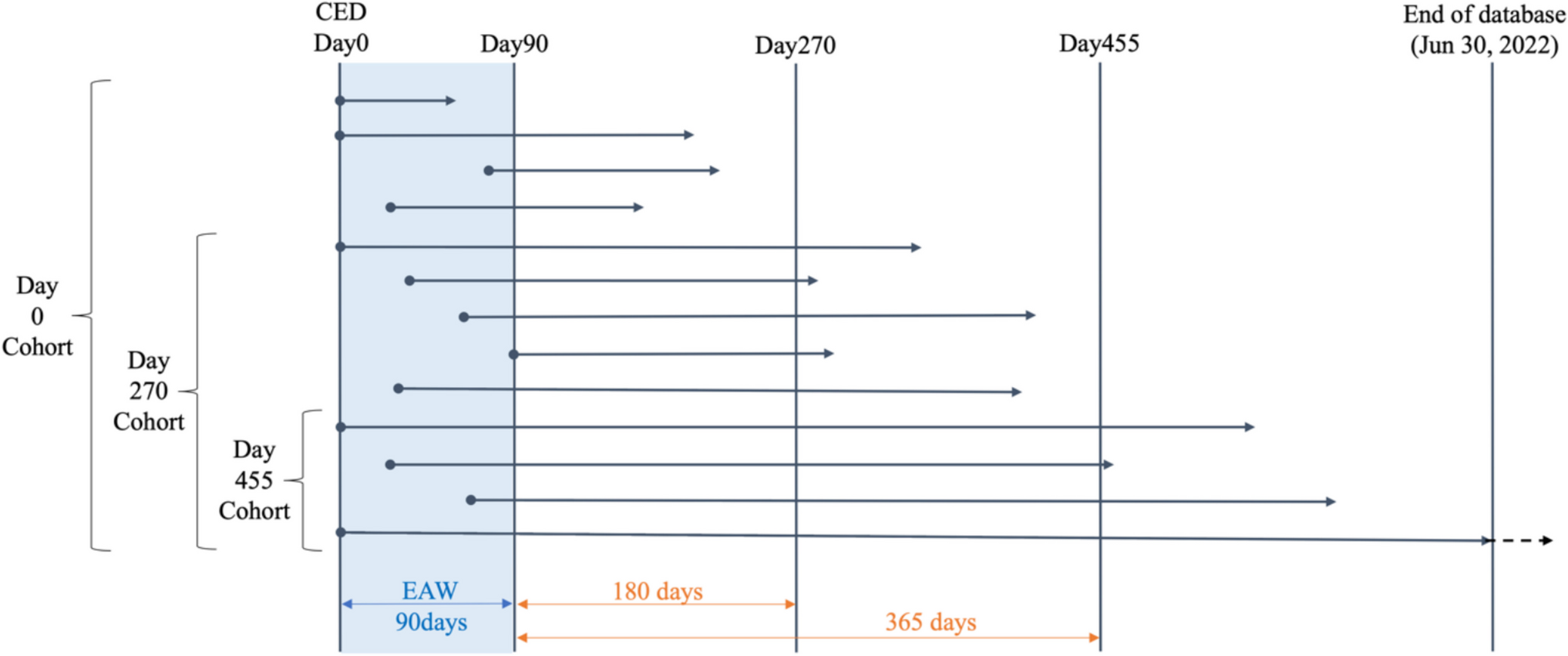
Accumulation of AGEs and imbalance of bone remodelingUnder hyperglycemic conditions, AGEs gradually accumulate through non-enzymatic glycosylation and bind to proteins in the bone matrix (such as collagen), resulting in impaired function of these structural proteins [32]. This accumulated AGEs destroy the mechanical strength of bone tissue, increasing bone fragility and fracture risk [33]. AGEs not only physically weaken the bone matrix, but by binding to receptors (RAGE), they also activate inflammation and oxidative stress responses in the body, further exacerbating the imbalance of the bone reconstruction process [34, 35]. The accumulation of AGEs leads to accelerated bone resorption and inhibits the function of osteoblasts, resulting in reduced bone formation and a significant increase in the risk of osteoporosis [36, 37].
Inflammation and bone remodeling imbalanceChronic low-grade inflammation in diabetesDiabetes, especially type 2 diabetes, is often accompanied by chronic low-grade systemic inflammation, which plays an important role in the development of osteoporosis [38]. Inflammatory markers, such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), are significantly elevated in patients with diabetes. The long-term chronic inflammatory state affects the bone reconstruction process through multiple signaling pathways [39]. These inflammatory factors can directly inhibit the differentiation and function of osteoblasts and impair bone formation [40]. In addition, inflammation also increases osteoclast activity and accelerates bone resorption [41]. This inflammation-mediated activation of osteoclasts and inhibition of osteoblast function jointly promote bone loss and the exacerbation of osteoporosis [42, 43].
Effects of inflammation on bone metabolismIn the pathological context of diabetes, the inflammatory response exerts profound effects on bone metabolism in multiple ways. First, inflammatory factors such as TNF-α can activate osteoclasts through the nuclear factor-κB (NF-κB) signaling pathway and increase the speed and intensity of bone resorption [44]. On the other hand, inflammatory factors also inhibit the activity of the Wnt signaling pathway, which plays a key role in the proliferation and differentiation of osteoblasts [45]. When the Wnt pathway is inhibited, osteoblast production is reduced and bone formation is inhibited [46]. In addition, cytokines such as IL-6 can further aggravate bone resorption by promoting the differentiation of bone marrow stromal cells into osteoclast precursors [47]. It can be seen that chronic low-grade inflammation caused by diabetes exacerbates the imbalance between bone resorption and bone formation during bone remodeling.
The role of oxidative stress in inflammation and bone remodeling imbalanceOxidative stress is a common accompaniment of inflammation and a common problem in patients with diabetes [48]. Under hyperglycemia, the production of reactive oxygen species (ROS) increases significantly, and ROS further promotes the inflammatory response through multiple signaling pathways, forming a vicious cycle [49]. Oxidative stress not only directly damages the functions of osteoblasts and osteoclasts, but also aggravates bone matrix damage by enhancing the accumulation of AGEs [50]. In addition, oxidative stress can activate pro-inflammatory signaling pathways such as NF-κB, further promoting the release of inflammatory factors, leading to increased osteoclast activity and accelerated bone resorption [51]. In contrast, osteoblast function is impaired in an oxidative stress environment, bone formation ability decreases, and the bone reconstruction process becomes more imbalanced. This imbalance in bone remodeling, driven by both inflammation and oxidative stress, is one of the important mechanisms of diabetes-related osteoporosis (Fig. 1).
Fig. 1
Mechanism of osteoporosis in diabetic patients
Bone remodeling imbalance mechanismHistomorphometric evidenceIn diabetes-related bone metabolism research, bone histomorphometry serves as the gold standard for directly evaluating bone formation and resorption at the tissue level, and has revealed characteristic abnormalities in bone remodeling. Studies have shown that patients with type 2 diabetes commonly exhibit reduced bone formation, decreased osteoblast surface coverage, and delayed bone mineralization, while osteoclast activity is not significantly increased. This low bone turnover pattern suggests that diabetic bone fragility is primarily driven by impaired bone formation, rather than by enhanced bone resorption as traditionally assumed. Compared with serum bone turnover markers or bone mineral density, histomorphometric findings more accurately reflect dynamic changes in bone remodeling and microstructural quality, providing important supplemental value for mechanistic research and clinical assessment. Therefore, in evaluating bone remodeling imbalance in diabetes, histological evidence should be considered, to avoid overemphasizing osteoclast activity while overlooking the central role of impaired osteoblast function in the development of osteoporosis [52, 53].
Osteoclast activation and bone resorptionThe bone reconstruction process in diabetic patients is affected by multiple mechanisms, and abnormal activation of osteoclasts is one of the key factors [54]. Osteoclasts are cells responsible for bone resorption and play an important role in bone metabolic balance [55]. In diabetes, osteoclast activity is significantly enhanced due to the effects of insulin resistance, hyperglycemia, and inflammatory factors (such as TNF-α, IL-6). These inflammatory factors directly promote the differentiation and activation of osteoclasts by activating pro-inflammatory signaling pathways such as nuclear factor-κB (NF-κB), leading to accelerated bone resorption. At the same time, the accumulation of AGEs further activates the function of osteoclasts by combining with RAGE, greatly increasing the rate of bone tissue loss. This ongoing bone resorption process is one of the main causes of imbalanced bone remodeling in patients with diabetes, ultimately leading to the development of osteoporosis [56]. In addition to the enhanced activity of osteoclasts, the suppression of bone formation is also an important component of diabetic bone fragility. One of the key mechanisms underlying reduced bone formation is the increased expression of sclerostin, a glycoprotein secreted by osteocytes that antagonizes the Wnt/β-catenin signaling pathway and inhibits osteoblast differentiation. Elevated sclerostin levels have been consistently observed in diabetic individuals and are associated with reduced bone formation activity [57]. Moreover, an increase in bone marrow adipocytes is another potential factor impairing osteogenesis. The accumulation of adipocytes in the bone marrow niche may suppress osteoblast development by altering mesenchymal stem cell differentiation, shifting it from the osteogenic to the adipogenic lineage. This change in lineage commitment contributes to reduced bone formation and diminished bone quality in patients with diabetes [58, 59].
Osteoblast dysfunction and reduced bone formationIn contrast to osteoclast activation, osteoblast function is significantly inhibited in diabetes [60]. Osteoblasts are the main cells for bone formation, and their proliferation and differentiation are regulated by multiple signaling pathways, such as the Wnt/β-catenin signaling pathway [61]. However, in diabetic patients, under conditions of hyperglycemia and chronic inflammation, the activity of the Wnt signaling pathway is significantly inhibited, leading to osteoblast dysfunction [62]. The production of osteoblasts is reduced, and the synthesis ability of bone matrix is also significantly reduced. In addition, insulin resistance prevents osteoblasts from responding properly to insulin signals, further exacerbating the reduced bone formation. Long-term low osteogenesis function directly leads to the decline of bone quality and increases the risk of osteoporosis [63].
GIP, GLP-1, and DPP-4 in bone metabolismIn addition to classical regulators such as inflammatory cytokines and AGEs, recent studies have highlighted the role of the pancreatic endocrine–bone axis in skeletal homeostasis, especially involving incretin-related pathways. Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are two major incretin hormones known to influence not only glucose metabolism but also bone remodeling. GIP receptors are expressed on both osteoblasts and osteoclasts, and GIP has been shown to promote osteoblast differentiation while suppressing osteoclast activity and bone resorption [64, 65]. Similarly, GLP-1 may exert osteoprotective effects via indirect mechanisms—such as enhancing insulin signaling and reducing systemic inflammation—and possibly through direct GLP-1 receptor activation on bone cells [66, 67]. Dipeptidyl peptidase-4 (DPP-4) is an enzyme that rapidly degrades both GIP and GLP-1. Inhibition of DPP-4, a common therapeutic approach in type 2 diabetes, increases circulating levels of active incretins and has been associated with improved bone turnover markers and a potential reduction in fracture risk in some studies [68]. Although the exact mechanisms remain under investigation, current evidence suggests that the incretin axis modulates bone remodeling through enhanced osteoblastogenesis, inhibition of osteoclastogenesis, and maintenance of bone mass and microarchitecture. These findings indicate that endocrine-metabolic signaling pathways, particularly the DPP-4/GIP/GLP-1 system, may be important contributors to the pathophysiology of diabetic bone fragility.
Changes in bone marrow microenvironmentDiabetes not only affects bone remodeling by directly acting on osteoclasts and osteoblasts, but also further disrupts bone metabolic balance by changing the bone marrow microenvironment [69]. Under normal circumstances, there are a series of signaling molecules and cytokines that regulate bone metabolism in the bone marrow microenvironment, such as bone morphogenetic proteins (BMPs) and chemokines (CXCLs) [70]. These molecules maintain the balance between bone resorption and bone formation. However, in patients with diabetes, the bone marrow microenvironment is significantly altered due to the effects of hyperglycemia, oxidative stress, and AGEs. The continued presence of pro-inflammatory factors destroys the normal bone marrow microenvironment, leading to impaired differentiation and function of osteoblasts, while promoting increased activity of osteoclasts [71]. In addition, diabetes also impairs bone-forming potential by affecting hematopoietic and mesenchymal stem cells in the bone marrow. This change in the microenvironment accelerates the progression of osteoporosis [72, 73].
Potential therapeutic approachesAnti-inflammatory therapyDue to the key role of chronic low-grade inflammation in diabetic osteoporosis, anti-inflammatory therapy is considered to be one of the potential effective strategies [74]. Studies have shown that pro-inflammatory factors such as TNF-α and IL-6 cannot only promote osteoclast activation, but also inhibit osteoblast differentiation [75]. Therefore, inhibiting the activity of these inflammatory factors may help slow bone loss. Nonsteroidal anti-inflammatory drugs (NSAIDs) have a certain effect in inhibiting inflammatory responses, but long-term use may be accompanied by side effects [76]. A more ideal solution is to develop biological agents targeting inflammatory factors, such as TNF-α inhibitors and IL-6 inhibitors, which are expected to restore the balance of bone remodeling by reducing the level of inflammation [77, 78]. In addition, antioxidants are also believed to be able to slow the progression of osteoporosis by reducing oxidative stress and inflammatory responses [79].
Application of bone remodeling promotersIn view of the problem of reduced bone formation in diabetes, the application of bone remodeling promoters provides an important way to prevent and treat diabetic osteoporosis [80]. Bisphosphonates have been widely used in the treatment of osteoporosis by inhibiting the activity of osteoclasts, which can significantly reduce the risk of fractures [81]. In addition, RANKL inhibitors such as denosumab also show significant inhibitory effects on osteoclast activity. This type of drug slows down bone resorption by blocking the RANKL signaling pathway and reducing the formation of osteoclasts [82]. For drugs that promote bone formation, recombinant parathyroid hormone (PTH) such as teriparatide can effectively stimulate the proliferation and differentiation of osteoblasts, increase bone mass and bone density [83]. Through the rational use of bone remodeling promoters, bone resorption and bone formation can be effectively balanced, reducing the risk of osteoporosis in diabetic patients.
New therapeutic targetsWith the in-depth study of the mechanism of diabetes-related osteoporosis, some new therapeutic targets have been gradually discovered and shown potential [84]. The accumulation of AGEs is considered to be an important driving factor for osteoporosis in diabetic patients, so the development of AGEs inhibitors has become a promising treatment direction. AGEs inhibitors such as aminoguanidine and ALT-711 can improve the structure and function of bone matrix by inhibiting the formation of AGEs or degrading the already formed AGEs [85]. In addition, anti-RAGE drugs are expected to reduce inflammation and oxidative stress by blocking the binding of AGEs to their receptors, thereby protecting bone tissue [86]. Another potential therapeutic target is oxidative stress regulators, which can indirectly slow down osteoclast activation and osteoblast dysfunction by reducing oxidative stress levels in diabetes [87]. The development of new targeted drugs is expected to provide more precise and effective solutions for the treatment of diabetes-related osteoporosis (Fig. 2). However, although the pathological role of AGEs in bone fragility has been well established, clinical evidence demonstrating that inhibition of AGEs accumulation directly improves bone mineral density (BMD) or reduces fracture risk remains limited [88]. Current clinical studies evaluating the efficacy of AGE inhibitors, such as ALT-711, are scarce and have not yet yielded conclusive results. Therefore, further clinical investigations are needed to determine whether targeting AGEs can translate into measurable benefits in fracture prevention and bone health improvement.
Fig. 2
Potential therapeutic approaches
Effects of antidiabetic medications on bone metabolismIn addition to glycemic control, several commonly used antidiabetic agents have been found to exert regulatory effects on bone metabolism, partly through modulation of AGEs, which are implicated in diabetic bone fragility. Among these, metformin has received particular attention for its potential to enhance osteoblast differentiation and inhibit osteoclast activity, primarily via activation of the AMPK signaling pathway [89, 90]. Furthermore, emerging experimental and clinical data suggest that metformin may reduce AGE accumulation through improved glycemic control, antioxidative capacity, and anti-inflammatory mechanisms, which may contribute to its observed association with lower fracture risk and increased BMD at multiple skeletal sites such as the lumbar spine and femoral neck. Pioglitazone, a thiazolidinedione and PPARγ agonist, has shown similar effects in reducing AGE formation, but its role in bone health remains controversial [91, 92]. While it may exhibit anti-inflammatory and glycemic benefits, clinical studies have reported increased fracture risk with long-term use, especially in postmenopausal women [93]. GLP-1-receptor agonists (GLP-1RAs), such as semaglutide and dulaglutide, originally developed for glucose regulation, have shown potential to affect both bone metabolism and AGE-related pathways [
Comments (0)