
Remember me

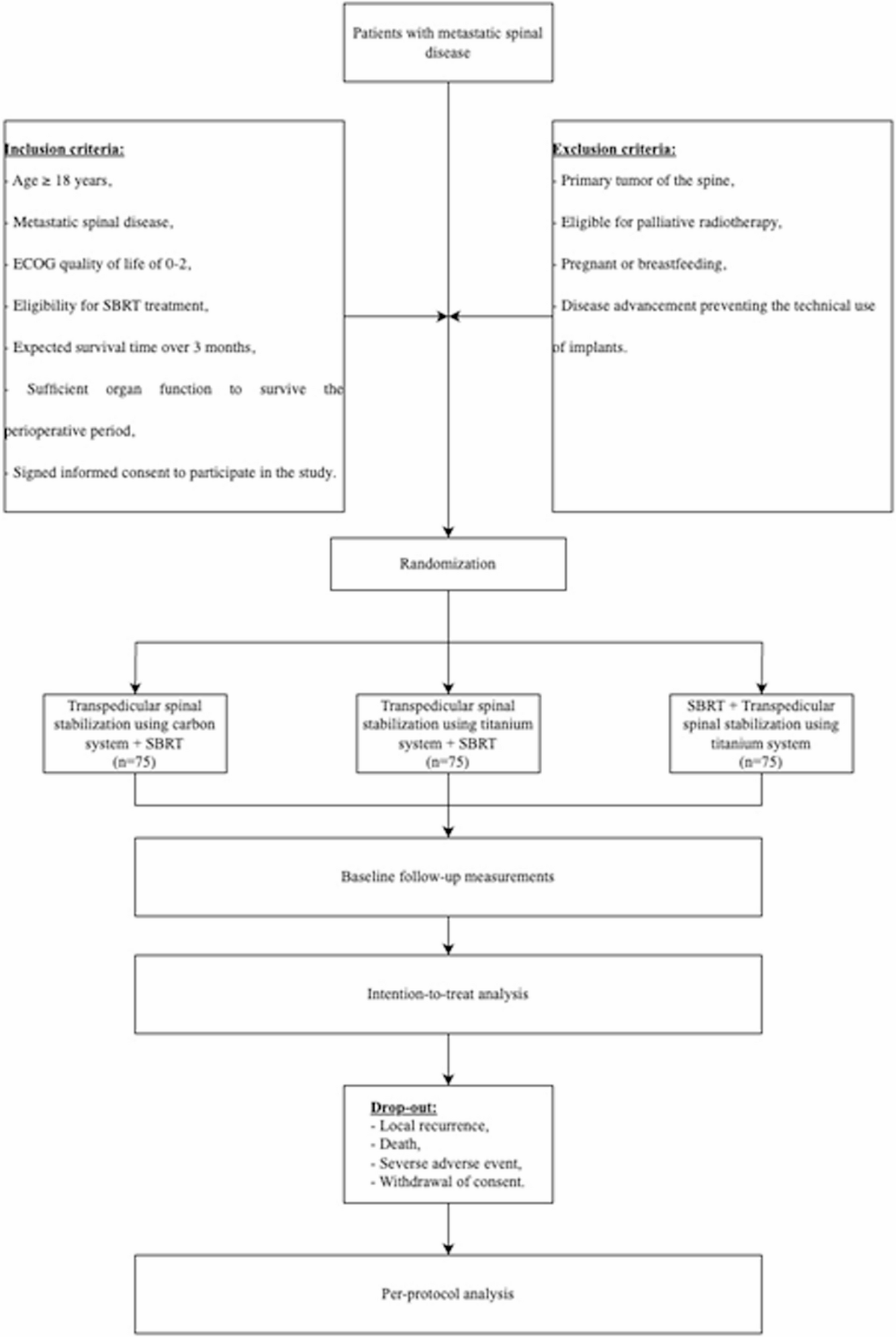
The CARBO-METASPINE trial is a randomized, non-blinded, three-arm clinical trial designed to evaluate the effectiveness of carbon fiber-based spinal stabilization systems in patients with metastatic spinal disease. This trial is conducted in compliance with the Helsinki Declaration and has received approval from the Bioethics Committee at the Polish Mother Health Center Institute in Lodz (approval no. 90/2022 from 20.09.2022, with extensions no. 69/2024 from 18.06.2024 and no. 53/2025 from 17.06.2025). The study is registered at Clinicaltrials.gov under the identifier NCT06293157 and is funded by the Polish Medical Research Agency (grant no. 2023/ABM/01/00013). There was no sponsor involvement in the study design, and sponsors will have no access to the randomization code or the database generated during the trial.
Study design and populationA total of 226 patients diagnosed with metastatic spinal disease will be recruited and randomly allocated into three groups in a 1:1:1 ratio using a permuted block randomization procedure (Fig. 1). The three arms of the study include:
Fig. 1
Flowchart of participants according to the Standard Protocol Items: Recommendations for Interventional Trials 2013 Statement. ECOG, Eastern Cooperative Oncology Group; SBRT, Stereotactic Body Radiotherapy
Arm I (Intervention Group, n = 76): Patients will undergo posterior (pedicle) stabilization using a radiolucent composite system (carbon fiber/PEEK), combined with stereotactic body radiation therapy (SBRT) delivered at 5 × 5 Gy (total 25 Gy).
Arm II (Control Group, n = 76): Patients will receive posterior stabilization using a titanium system, followed by the same SBRT regimen (5 × 5 Gy, total 25 Gy).
Arm III (Control Group, n = 75): Patients will receive preoperative SBRT (5 × 5 Gy, total 25 Gy) followed by posterior stabilization using a titanium system.
Arm II was chosen as a comparator because it represents the current standard treatment protocol for metastatic spinal disease. Arm III serves as a control for the alternative treatment paradigm, providing a basis for SBRT planning without the use of fixation systems.
Specific objectives related to benefits and harmsBenefits of Carbon Fiber Implants:
Enhanced radiotherapy precision: Assess the impact of carbon fiber-based implants on planning and dose distribution in SBRT.
Enhanced local tumor control: Assess whether carbon fiber implants contribute to improved PFS compared to traditional titanium implants.
Reduction of imaging artifacts: Investigate the extent to which carbon fiber implants reduce radiological artifacts, improving postoperative imaging quality.
Harms and ComplicationsSurgical complications: Monitor for adverse events related to the surgical application of carbon fiber implants, including infection, hardware failure, and neurological deterioration.
Post-radiotherapy side effects: Track the occurrence of complications after SBRT, such as tissue damage, spinal cord injury, and other radiation-related adverse effects, in both the experimental and control groups.
Risk of recurrence: Determine if the use of carbon fiber implants affects the timing and ability to detect local recurrence of metastatic spinal disease.
Randomization and allocation concealmentPatients will be randomly assigned to the study arms using permuted block randomization, a method that ensures an equal distribution of participants across the groups throughout the trial. This approach helps maintain balance between the treatment arms, minimizing the risk of allocation bias. To further reduce bias and protect the validity of the study, allocation concealment will be rigorously maintained throughout the entire recruitment and treatment process. Both patients and researchers will be informed about the allocation due to the nature of the intervention. Since the study involves different surgical stabilization methods (carbon fiber vs. titanium implants), it is not possible to blind either the participants or the treating clinicians to the group assignment. However, allocation concealment will still be maintained up until the point of randomization to minimize bias before the intervention. Despite the lack of blinding, efforts will be made to ensure objective outcome assessments and reduce any influence of the knowledge of group allocation on the evaluation of clinical results. All statistical analyses will be performed by an independent biostatistician who will remain blinded to group allocation throughout the study. The randomisation code will be unmasked for this analyst only after the final database lock and completion of the primary analysis.
The personnel responsible for enrolling participants will not have access to the randomization sequence. Enrollment staff will input each eligible participant’s details into a secure, web-based randomization system. The randomization algorithm (permuted blocks) is stored centrally on a password-protected server, and the sequence is only released after the data are submitted.
The designated study coordinator (or operating surgeon) who performs the assigned intervention will learn the allocation only after the system provides it; they also have no prior knowledge of the sequence itself or upcoming assignments. Therefore, both enrollment staff and intervention-assigning staff remain blinded to the random allocation list, ensuring complete allocation concealment until the moment of assignment.
Additionally, to preserve the integrity of the outcome analysis, data analysts will be blinded to the group assignments. This ensures that their analysis remains unbiased and that the results are not influenced by prior knowledge of the treatment groupings.
Study sitesThe trial was initially launched at the Copernicus Memorial Hospital in Lodz, Poland, within the Department of Neurosurgery and Neurooncology. Two additional recruiting centers have received approval from the Institutional Board: Jan Biziel University Hospital No. 2 in Bydgoszcz, Poland, and the Lower Silesian Centre for Oncology, Pulmonology, and Hematology in Wroclaw, Poland. As more recruitment centers are added, the Department of Neurosurgery and Neurooncology at Copernicus Memorial Hospital will continue to be the primary study site.
Steering committeeThe Steering Committee (SC) will be led by KK (neurosurgeon, PhD). The SC will consist of ŁK (radiation oncologist, PhD), MT (neurosurgeon, PhD), BP, AK, MS, KR (all medical doctors), JF, MP (radiation oncologists), KD, FA (medical students), KZ (psychologist), and MJO (biostatistician, PhD). The SC will oversee the study’s progress, addressing decisions related to participant recruitment, retention, and study dropouts.
Protocol developmentThe study protocol was developed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) [11]. The protocol adheres to established guidelines for the design, conduct, and reporting of clinical trials, ensuring a rigorous and transparent methodology throughout the study.
RecruitmentPatients with spinal metastases will be recruited from oncological and neurosurgical centers. A biopsy is not mandatory if the radiologic appearance and clinical history conclusively indicate metastatic disease. In cases where the diagnosis is uncertain, a biopsy may be conducted.
Screening and Study QualificationPatient screening and qualification will involve the following procedures:
Radiological Imaging:
Up to one month prior to treatment qualification, a contrast-enhanced MRI of the spine, or a contrast-enhanced CT scan (if MRI is contraindicated), will be performed.
An appropriate imaging modality (contrast-enhanced MRI or CT) will be performed 2 weeks prior to treatment.
Laboratory Tests:
A complete blood count, electrolyte panel, coagulation profile, glucose levels, renal function tests, albumin, total protein, and alkaline phosphatase will be performed one day prior to treatment.
Screening for hepatotropic viruses (Hepatitis B and C) and determination of blood type, conducted within three days before treatment.
Clinical and Neurological Evaluation:
Assessment of general condition and neurological functions during patient enrollment.
Quality of life assessment using the ECOG scale and patient-reported outcomes before and after the surgical procedure.
Demographics and Medical History:
Data such as age, sex, primary oncological diagnosis, and comorbidities will be collected upon patient admission for the procedure.
Eligibility CriteriaInclusion Criteria:
Metastatic spinal disease,
ECOG quality of life of 0–2,
Eligibility for SBRT treatment [12],
Expected survival time > 3 months,
Signed informed consent to participate in the study,
Sufficient organ capacity allowing to survive the perioperative period.
Exclusion Criteria:
Primary tumor of the spine,
Age < 18 years old,
Expected survival time < 3 months,
Eligibility for palliative radiotherapy,
No informed consent to participate in the study,
Pregnancy or breastfeeding,
The advancement of the disease preventing the technical use of implants.
All eligible patients will be offered the opportunity to participate in the trial. After the neurosurgeon explains the study design and intervention, patients will confirm their intention to participate by signing the written informed consent form (ICF). The participant flowchart is shown in Fig. 1.
Patients who do not meet all inclusion criteria, meet any exclusion criteria, or withdraw consent will receive standard care for spinal metastatic disease. Reasons for screening failures will be documented.
Definition of study completion and early terminationStudy completion is defined as the point when all data has been entered and all queries resolved. Interim analyses are planned once 50% and 75% of patients have been enrolled. If the study’s statistical assumptions are met at these interim points, the trial may be terminated before the planned completion date.
Informed consentParticipants must personally sign and date the approved version of the informed consent form before any procedures are performed. Both written and oral versions of the participant information and consent form will be provided, ensuring that at least the following details are included: a comprehensive study description, implications for the participant, protocol limitations, known adverse effects, and associated risks. It will be explicitly stated that the participant may withdraw from the study at any time without affecting future care or rights, and without the need to justify the decision. The participant will be given adequate time to review the information and ask questions before deciding to participate. Written informed consent will be obtained based on the participant’s dated signature and the signature of the individual providing the information and obtaining consent. The person responsible for obtaining consent must have the appropriate qualifications, experience, and authorization from the principal investigator. The participant will receive a copy of the signed informed consent form, and the original signed document will be kept at the research center.
Follow-Up assessmentsFollow-up will continue for 5 years post-surgery and will be terminated with patient death or local recurrence of the disease. The follow-up will include the following procedures:
Neurological assessmentOn the day of surgery and subsequent days during hospitalization to monitor for acute complications.
ImagingContrast-enhanced MRI or CT within three days post-intervention to assess tumor status, stabilization efficacy, and potential complications.
Scheduled follow-up appointments every three months post-radiotherapy or surgery (depending on the study arm) to assess long-term outcomes, local recurrence, and device stability (Fig. 2).
Fig. 2
Schedule of enrolment, surgical and radiotherapy interventions, and time-point assessments
Radiological assessments to monitor for tumor progression or recurrence (increase in tumor volume or new contrast enhancement) every three months.
>Adverse eventsDocumentation of any surgical or radiotherapy-related adverse events according to the National Cancer Institute Common Toxicity Criteria (NCI CTC).
Postoperative wound evaluation.
Quality of Life and Pain Assessment:
Follow-Up assessments Periodic quality-of-life assessments and pain scoring using the Visual Analog Scale (NRS) will be performed.
InterventionsThe radiotherapy standards outlined in this protocol follow guidelines from the European Society for Radiotherapy and Oncology (ESTRO) [13] and the American Society for Radiation Oncology (ASTRO) Clinical Practice Guidelines. Surgical procedures adhere to recommendations from the Polish Society of Spinal Surgery [10]. Radiotherapy target volumes for preoperative and postoperative spine lesions were delineated according to the contouring atlases by Cox et al. and Redmond et al. [14, 15]
Surgical ProceduresInstrumented spinal stabilization using pedicle screws may involve the use of vertebral body prostheses. The timing of the intervention depends on the patient’s qualification pathway:
Acute or Rapidly Progressing Neurological Deficits: In these cases, the neurosurgical team will quickly assess the patient, and urgent decompression of neural structures will be performed. Stabilization procedures may be carried out during the same operation or postponed, depending on the patient’s clinical status and intraoperative findings.
Elective Cases: For elective surgeries, patient eligibility will be determined in an oncological multidisciplinary conference involving a clinical oncologist, radiation oncologist, and spine surgeon. Following consensus, appropriate treatment will be carried out within 14 days. For operative cases, stereotactic radiotherapy will be administered between 14 and 21 days after discharge from the Neurosurgery Department.
Surgical TechniquesDecompression of Neural Structures:
Indirect decompression, commonly performed via laminectomy, involves removing the lamina above the area of spinal cord compression. The procedure may be part of a multistep approach and is tailored to the patient’s condition.
Separative (Separation) Surgery:
Described by Angelov and Benzel [16], this technique involves partial resection of the tumor adjacent to neural structures to create conditions favorable for stereotactic radiotherapy. This approach combines indirect decompression and typically requires a posterolateral approach to the dural sac. Separative procedures are often combined with instrumented stabilization, and the dural sac should maintain approximately 2 mm of clearance from the tumor.
Piecemeal Corpectomy:
In cases where the spine surgeon opts for more extensive tumor removal, a piecemeal corpectomy involves removing the entire infiltrated vertebral body in fragments. Reconstruction of the anterior spinal column with a vertebral body prosthesis is required.
En-bloc Corpectomy:
Rarely, an en-bloc corpectomy may be performed, where the entire vertebral body is removed intact. This involves isolating the vertebral body from the adjacent ligaments, intervertebral discs, and posterior spinal elements, with anterior column reconstruction and pedicle screw stabilization following the procedure.
Stereotactic Body Radiotherapy (SBRT)A five-fraction SBRT regimen will be employed: 5 × 5 Gy (total 25 Gy). The following areas will be contoured by a radiation oncologist:
Gross Tumor Volume (GTV).
Clinical Target Volume (CTV).
Planning Target Volume (PTV).
The PTV will be the CTV with a 2 mm margin (which can be individually adjusted), excluding the spinal cord area, which will have a Planning Risk Volume (PRV) margin. GTV, CTV, and PTV contours will be based on the anatomical recommendations from the International Spine Radiosurgery Consortium (ISRC), as described by Cox et al. [14], Redmond et al. [15], and Dunne et al. [17].
For eligible patients, the CTV area will be maintained 1–2 mm from the PRV. The GTV, CTV, PTV, and OAR areas will be audited by a radiation oncologist in collaboration with a medical physicist based on the following guidelines:
Coverage parameters included the percentage of the Clinical Target Volume (CTV) and Planning Target Volume (PTV) that received at least 95%, 98%, and 100% of the prescribed dose (V95%, V98%, and V100%, respectively), as well as the maximum dose within the PTV.
Spinal Cord Protection: Priority must be given to spinal cord protection, potentially compromising PTV (CTV) coverage.
Dose distributionAt least 90% of the PTV volume should receive 100% of the planned dose (PD).
Over 80% of the CTV should receive 100% of the PD.
Minimum doseThe minimum dose (Dmin) in the GTV should be 25 Gy (for the five-fraction regimen).
Surgical procedures Optimal Conformity Index (CI): Ideally, CI should be < 1.2.
OutcomesPrimary outcomesThe primary objective of the study is to evaluate the time to and frequency of local recurrence (Progression-Free Survival, PFS) following stereotactic body radiotherapy (SBRT). Local recurrence will be defined according to radiological imaging, using either contrast-enhanced MRI or, if MRI is contraindicated, contrast-enhanced CT. Local recurrence will be identified as an increase in tumor volume or the appearance of new contrast enhancement after contrast administration.
The primary endpoint will be evaluated using the log-rank test and Cox proportional hazards regression model
Secondary outcomeSecondary outcomes of the study include:
Evaluation of the DICE similarity coefficient between the treatment groups.
Assessment of subjective difficulty in radiotherapy treatment planning.
Frequency of post-radiotherapy complications, evaluated according to the National Cancer Institute Common Toxicity Criteria (NCI CTC).
Failure rate of stabilization systems used in surgical stabilization.
Pain assessment using the Visual Analog Scale (NRS) across all study groups.
Frequency of postoperative infectionsOutcomes Differences in dosimetric parameters, including Dmin PTV/CTV, D95, D98, D100, conformity index (CI), and homogeneity index (HI).
InterventionsInterventions will be performed within 14 days of the oncological consensus meeting for elective cases, or as soon as possible for urgent neurosurgical interventions. Radiotherapy for Arms I and II will be administered within 14–21 days after surgery.
To maintain consistency across the study arms, all surgical procedures will be performed following standardized protocols that unify the surgical technique. Radiotherapy planning and delivery will adhere to established guidelines [15, 16], and dosimetric parameters will be recorded for further analysis.
Data collection, management, and analysisAll clinical data collected will be anonymized at the time of data entry, with personal identifiers encoded. Study data will be stored on password-protected external drives and servers accessible only to the study investigators. Publication of study results will ensure complete patient anonymity, and no personal data will be identifiable.
Data will be processed in accordance with Article 13(1) and (2) of the European Parliament and Council Regulation (EU) 2016/679 of 27 April 2016, regarding the protection of natural persons in relation to the processing of personal data and the free movement of such information.
Source data and data accessSource documents refer to the original media on which data are recorded and from which participants’ data are subsequently transferred into the Clinical Report Forms (CRFs). These include, but are not limited to, hospital records (which provide information on medical history and concurrently or previously administered medications), clinical and hospital files, laboratory and pharmacy documentation, diaries, microfilms, imaging results, and correspondence. Entries in the CRF will be considered source data if the CRF serves as the primary record (e.g., in the absence of any other written or electronic record). All documents will be stored securely and maintained with confidentiality. Except for the signed informed consent form, participants will be identified by a number/code rather than by name in all study documents. Direct access will be granted only to authorized representatives of the sponsor, the receiving institution, and regulatory authorities for the purpose of monitoring, auditing, and inspecting the study.
Data collection and documentation storageClinical data will first be recorded in paper CRFs that carry only study-specific identification codes, ensuring participant anonymity. Trained study coordinators will subsequently transcribe these data into an electronic eCRF housed on a password-protected server. Access to the eCRF is role-restricted and granted solely to the principal investigator, the study biostatistician, and designated coordinators.
Data collection, management, and analysis All imaging and laboratory analyses will be conducted in accredited, standardized facilities that operate under validated protocols and adhere to external quality assurance schemes. Radiological and clinical rating scales (e.g., ESCC, SINS, Frankel) will be scored by qualified investigators; each assessment will be reviewed and countersigned by the principal investigator, who has overall responsibility for data integrity and protocol compliance.
Clinical report formStudy data will be collected using both paper and electronic CRFs. A separate CRF will be maintained for each enrolled participant and will be continuously updated to ensure that the data reflect the participant’s current status at each phase of the study. The CRF will not include personally identifying information, such as the participant’s name, initials, or date of birth. Identification will be carried out using a code, for example, combining the participant number with the randomization code. Only the principal investigator, study coordinator, and medical biostatistician are permitted to edit the CRFs. Data collected through the CRFs will then be entered into an electronic database for analysis by the study coordinator. All critical variables—including primary and key secondary outcomes as well as adverse-event grades—will be entered independently by two coordinators (double data entry). Any discrepancies detected by the system will be automatically flagged and resolved through verification against the original source documents. In addition, embedded logic and range checks will block the entry of out-of-range, implausible, or internally inconsistent values, thereby safeguarding data integrity at the point of capture.
Risk assessmentThe study will be conducted in accordance with the currently approved protocol, the principles of Good Clinical Practice (GCP), applicable regulatory requirements, and standard operating procedures. Given the nature of this study, no significant risk is anticipated for the participants.
Monitoring, audits, and inspectionsWithin the hospital infrastructure, a dedicated Clinical Research Center (CRC) functions as an independent quality assurance unit responsible for auditing and monitoring all investigator-initiated and sponsored studies conducted on-site. Comprehensive audits are performed every six months and include verification of essential trial documentation and source data, assessment of recruitment metrics against projected timelines, and evaluation of protocol adherence, including informed-consent procedures and adverse event reporting. Each audit results in a written report with findings, graded by severity, and detailed recommendations or corrective actions. The CRC tracks the implementation of these recommendations until full resolution, ensuring continuous compliance with GCP and institutional policies across all ongoing trials.
Regular monitoring of the clinical trial will be performed. Data will be evaluated for compliance with the protocol and the source documents as defined in this document. Coordinators and authorized personnel will verify that the clinical trial, including data generation, documentation, and reporting, is conducted in accordance with the protocol, GCP, and relevant regulatory requirements.
Study documentation and source data/documents will be made available to auditors and inspectors. During inspections, any queries will be promptly clarified. All parties involved are required to maintain strict confidentiality regarding participant data.
Confidentiality of personal dataDirect access to source documents will only be permitted for monitoring, audits, and inspections. The research team will have access to the protocol, but the randomization sequence will be restricted solely to the coordinator at the main center and the medical biostatistician.
Harms definition and assessmentHarms will be defined as any adverse events or complications that occur during or after the intervention, potentially resulting from the surgical procedure, the use of the stabilization implants, or radiotherapy. These may include, but are not limited to, surgical site infections, hardware failure, neurological deterioration, radiation-induced injuries, or other treatment-related side effects.
Harms will be systematically assessed throughout the study. All participants will be monitored closely during the postoperative period and follow-up visits. Adverse events will be documented according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE). These events will be evaluated in terms of severity, causality, and the need for intervention.
Additionally, a non-systematic assessment will be conducted, where participants will be encouraged to report any side effects or complications that arise outside of regular follow-up visits. This will be done through patient-reported outcome measures (PROMs) and direct communication with the study team. Regular assessments will be conducted at predefined intervals to ensure early detection and proper management of any harm, with all findings reported promptly to the ethics committee and regulatory authorities.
Participant flow coordination and follow-up proceduresClinical report form Study coordinators will actively manage patient transfers between the Neurosurgery Department and Radiotherapy Units through structured telephone communication. Coordinators will schedule outpatient appointments, relay detailed information about treatment plans and visit schedules, and document all interactions in the electronic case-report system. If a participant fails to respond or does not attend a scheduled visit, a second telephone contact attempt will be made within 48 h. Continued non-compliance will result in the issuance of a formal written summons, and, if necessary, outreach to designated family members to facilitate re-engagement and ensure continuity of follow-up.
Deviations from the protocol and serious violationsA deviation is defined as any departure from the ethically approved content of the study protocol, any other study document, or any research process (e.g., the consent procedure or administration of the investigational product), from GCP, or applicable regulatory criteria. All protocol deviations will be documented on a protocol deviation form and appended to the primary study documentation.
Deviations from the protocol and serious violations Any essential modifications to the protocol will be communicated promptly to all relevant parties, including study participants, the ethics committee, regulatory authorities, and the sponsor. These modifications will be documented and submitted for approval to the relevant regulatory bodies and ethics committees prior to implementation. The principal investigator and study team will ensure that all involved centers and investigators are informed of any changes through written notifications. If necessary, updated informed consent forms will be provided to participants, explaining the nature of the modifications and obtaining re-consent if required. Additionally, any modifications will be logged in the trial’s records, and the changes will be communicated through official study meetings and documentation to ensure transparency and compliance with ethical standards.
StatisticsBelow is a detailed description of the planned statistical analysis for the study. No separate statistical analysis plan will be utilized.
Statistical analysis planThe evaluation of time to and frequency of PFSx following stereotactic body radiotherapy (SBRT) will be performed using the log-rank test and Cox proportional hazards regression model. Additional analyses will include both parametric and non-parametric tests, depending on the distribution of the variables under investigation.
For quantitative variables that are normally distributed, a Student’s t-test will be employed for two-group comparisons or ANOVA for comparisons among three or more groups, followed by post hoc Student’s t-tests.
For non-normally distributed quantitative variables, the Mann–Whitney–Wilcoxon test will be applied for two groups or the Kruskal–Wallis test for three or more groups, followed by post hoc Wilcoxon tests.
Where justified, Bonferroni correction will be applied to account for multiple hypothesis testing.
For qualitative variables, either the chi-square test or Fisher’s exact test will be used, depending on expected frequencies.
Additionally, Cohen’s kappa coefficient will be calculated to assess inter-rater agreement.
For exploratory purposes, algorithms such as Principal Component Analysis (PCA) and its modifications, capable of analyzing categorical or mixed data (e.g., Correspondence Analysis [CA], Multiple Correspondence Analysis [MCA], and Multiple Factor Analysis [MFA]), will be utilized.
All statistical analyses will be conducted using R (version ≥ 4.2.0).
Hypothesis and significance LevelThe study hypothesis assumes that the use of carbon fiber-based spinal stabilization systems in metastatic spinal disease will facilitate simpler and more precise stereotactic radiotherapy planning, resulting in superior dosimetric parameters that enhance local disease control. The implementation of radiolucent implants with a low propensity for generating imaging artifacts is expected to improve the quality of post-treatment imaging follow-up and the assessment of local recurrence. The treatment is not expected to be associated with additional late adverse effects.
The significance level for the study is set at α = 0.05 with a testing power of 1-β = 0.8.
Sample size estimation and power calculationsThe sample size was estimated based on the assumed hazard ratio of 0.62 for local failure. A total of 226 patients provides 80% power to detect the difference at the two-sided α = 0.05 level, accounting for a 10% censoring rate. The researchers adjusted the statistical analysis assumptions based on the literature, lowering the median PFS from 2 years to 6 months and modifying the censoring rate to reflect a median survival in metastatic spinal disease of 19.5 months (adjusting the censoring rate from 0.3 to 0.1). Power calculations were performed using R v.4.0.2, based on Schoenfeld’s equation for the Cox proportional hazards model.
The primary strategy for maintaining the planned recruitment involves actively promoting the study within the medical community to ensure widespread awareness among healthcare professionals. This includes targeted outreach to oncologists, neurosurgeons, and radiologists who are involved in the treatment of metastatic spinal disease. Additionally, efforts are focused on expanding the network of recruiting centers, engaging new institutions that meet the study’s criteria for participation. By increasing the involvement of clinical centers and fostering collaboration with key stakeholders in the field, the study aims to enhance patient enrollment and ensure that recruitment targets are met within the established timeline.
Interim analysesInterim analyses will be conducted after 50% and 75% of patients have been enrolled. If the study’s statistical assumptions are met at any of these interim points, the trial may be terminated earlier. The principal investigator will have access to the interim analysis results and will decide whether to continue the study based on these findings.
Handling of missing dataMissing data will be addressed using appropriate statistical methods to minimize bias and ensure the robustness of the analysis. The approach to handling missing data will depend on the nature and extent of the missing information:
Descriptive Statistics: Before applying any imputation techniques, the extent and pattern of missing data will be assessed. Descriptive statistics will be used to evaluate the missing data at each time point and identify any systematic patterns.
Imputation Methods: If missing data is deemed to be missing at a rate of up to 10% randomly, multiple imputation techniques will be employed to estimate the missing values based on the observed data. This approach will help maintain statistical power and ensure valid inferences while preserving the variability in the dataset.
Complete Case Analysis: In cases where the proportion of missing data is minimal and does not significantly impact the dataset, a complete case analysis may be performed, where only participants with complete data for the variables of interest are included in the study.
Sensitivity Analysis: Sensitivity analyses will be conducted to assess the potential impact of missing data on the study results. This will involve comparing the results of the imputed data with those from the complete case analysis to ensure that missing data does not significantly impact the study’s conclusions.
Handling of Dropout Data: Participants lost to follow-up will be accounted for in the analysis, with dropouts carefully considered as part of the sensitivity analysis. If necessary, last observation carried forward or other methods for handling missing follow-up data may be considered, depending on the nature of the study design and data.
Additional plan analysisAdditional analyses will be performed to test the robustness and generalisability of the primary findings. Prespecified subgroup analyses will first explore potential effect modification by key baseline characteristics: patient age (< 65 vs. ≥ 65 years), sex, baseline ESCC grade (1c–2 vs. 3), number of spinal metastases (single vs. multiple), baseline ECOG performance status (0–1 vs. 2), and primary-tumour histopathology (breast, prostate, lung, kidney, myeloma/lymphoma, other). For each characteristic, an interaction term with treatment arm will be introduced into the primary Cox proportional-hazards model; a two-sided interaction p-value < 0.10 will be interpreted as evidence of heterogeneity of treatment effect.
A series of sensitivity analyses will then be conducted to address potential sources of bias. These will include: repeating the primary analysis after multiple imputation of missing covariates and outcomes under the missing-at-random assumption; a per-protocol analysis restricted to participants without significant deviations from their assigned intervention; a competing-risks regression treating non-spine-cancer deaths as competing events for local progression; and re-estimation of treatment effects using robust standard errors to account for clustering by recruiting centre.
Data managementAll clinical data will be anonymized by coding personal identifiers at the time the physician completes the questionnaire. The collected and coded clinical data will be stored on password-protected external drives and servers, accessible only to the research team. The publication of study findings will ensure complete anonymity of patients, with no possibility of identifying personal data. All data will be processed in accordance with Article 13(1) and (2) of the European Parliament and Council Regulation (EU) 2016/679 on the protection of natural persons regarding the processing of personal data and the free movement of such information.
Source Data and Data AccessStatistics Source documents are the original media on which data are recorded and from which data are transferred into the participant’s CRF. These documents include, but are not limited to, hospital records (used to complete CRFs regarding the patient’s medical history and concurrently or previously administered medications), clinical and hospital files, laboratory and pharmacy documentation, diaries, microfilms, imaging results, and correspondence. Entries in the CRF are considered source data if the CRF serves as the primary record (e.g., in the absence of any other written or electronic record). All documents will be stored securely and maintained in strict confidentiality.
Plans to Communicate Trial Results to Participants, Healthcare Professionals, the Public, and Other Relevant GroupsReporting in the Trial RegistryThe trial results will be reported in the clinical trial registry (ClinicalTrials.gov, NCT06293157) once the study is complete. This will include the final study outcomes, including primary and secondary endpoints, adverse events, and any other relevant findings. The registry will ensure that the study results are publicly accessible and transparent, in accordance with ethical guidelines and international standards for clinical trial reporting.
Communicating Results to Healthcare ProfessionalsThe findings will be shared with the medical and scientific community through the publication of the full trial results in peer-reviewed journals. These journals will focus on clinical oncology, neurosurgery, and radiotherapy, ensuring that relevant healthcare professionals are informed about the trial’s outcomes. In addition to peer-reviewed articles, results will be presented at national and international scientific conferences related to oncology, neurosurgery, and radiotherapy, to foster discussion and dissemination within the professional community.
Public Communication of ResultsA plain language summary of the study’s results will also be made available to the public through the trial registry, as well as through press releases and the institution’s website. This will ensure that the results are accessible to a broader audience, including patients, caregivers, and other members of the public interested in the trial’s outcomes. The public summary will highlight the impact of the findings on the treatment of metastatic spinal disease and provide context for how the results may influence future clinical practice.
EthicsThe study will be conducted in strict accordance with the protocol and the provisions of the current version of the Declaration of Helsinki, as well as the ICH GCP guidelines, and Polish law. Both the Ethics Committee and the regulatory authority will receive annual and periodic safety reports during the study’s conduct and will be promptly informed of any study discontinuation or termination in accordance with the regulations.
The principal investigator affirms and upholds the participant’s right to privacy and commits to complying with all applicable regulations regarding data protection. The medical information obtained from individual participants during the study will be treated as confidential and will not be disclosed to any third parties. For the purpose of data verification, authorized representatives of the sponsor, relevant regulatory authorities, or the ethics committee may request direct access to portions of the medical records related to the study, including the participants’ medical history. The principal investigator and the research team will inform participants about the use of their data, ensuring they fully understand the purpose and scope of the study. Additionally, they will obtain informed consent from each participant before collecting or using any data, in accordance with relevant ethical guidelines. All access to medical records and data will be conducted in strict compliance with confidentiality agreements and applicable laws regarding privacy.
Additional consent provisions will be provided to participants for the collection and use of their data and biological specimens, including blood and tissue samples, in ancillary studies, if applicable. Prior to enrollment, participants will be informed about the potential use of their medical records, diagnostic images, and biological samples for future research purposes, including biobanking. Separate consent will be obtained specifically for the biobanking of blood and tissue samples. Participants will receive detailed information regarding the storage, potential future uses, and long-term preservation of these specimens, which may include research on metastatic spinal disease, treatment outcomes, and the effects of the intervention. Participation in biobanking will be voluntary, and participants will be assured that they have the right to withdraw their consent for the use of these samples at any time, without affecting their participation in the main trial or access to standard care.
Plans to communicate trial results to participants, healthcare professionals, the public, and other relevant groups Substantial amendments to the protocol shall only be implemented after obtaining approval from the Bioethics Committee and the regulatory authorities. In situations where immediate deviations from the protocol are necessary to protect the rights, safety, and welfare of the participants, such deviations may be implemented without prior approval from the sponsor or the responsible authorities. These deviations must be documented and reported to the sponsor and the ethics committee/the appropriate regulatory authority as soon as possible. Any minor amendments must be reported to the appropriate authority promptly, if applicable, and to the Bioethics Committee as part of the annual safety report.
Conflict of InterestThe authors declare that they have no competing interests.
ApprovalsUpon sponsor approval, the protocol, the informed consent form, the patient information sheet, and all other materials/documents used in the study will be submitted to the appropriate Bioethics Committee, regulatory authorities, and receiving organizations for written approval. The investigator shall submit any substantive amendments to the original documents for review and, if necessary, obtain permission from the aforementioned entities prior to implementing those changes.
Carbon-fiber-reinforced implants have been available for several years; however, their role in the surgical treatment of metastatic spine disease remains uncertain [Khan]. Theoretically, radiolucent implants reduce artifacts generated during radiological examinations, potentially facilitating more accurate radiotherapy planning [18]. Furthermore, artifacts from commonly used titanium implants may increase dose degradation and affect the delivery of particles to the tumor. Lastly, radiological follow-up likely provides the opportunity to detect recurrence earlier in carbon-fiber instrumented patients. The authors performed a pair-matched analysis of the cases treated using carbon-fiber screws with titanium ones and showed similar PFS between the groups (143 days versus 214 days; p = 0.41); however, when the recurrence was present, it could be diagnosed earlier in the carbon-fiber group than in the titanium (94 days versus 189 days; p = 0.013) [19].
Recently, Hubertus et al. published a surgical cohort of 457 patients treated with carbon-fiber implants in spine oncology [20]. The authors presented a"real-life” scenario from four German centers. The study included both metastatic spinal disease, which was predominant, and non-metastatic infiltration, such as multiple myeloma and lymphoma. Generally, postoperative complications were reported in 13% of cases, primarily surgical site infections in 7%. Instrument-related problems were observed in 2% of cases, primarily concerning the screws. A relatively low percentage of cases were observed using MRI: 19% underwent direct postoperative MRI, and 27% were imaged during follow-up. Moreover, only 3% of patients underwent SBRT as a form of radiotherapy. The radiolucency of the carbon-fiber implants seems to influence mainly the stereotactic techniques of radiotherapy. The authors concluded that heterogeneity and lack of standardization between centers could bias the results [20]. From a surgical point of view, carbon-fiber implants are a valuable alternative to titanium ones, which was a conclusion from other reports [21, 22].
Despite several retrospective analyses, case studies, and biomechanical reports, there is a lack of high-quality data, particularly randomized controlled trials, regarding the effectiveness of carbon-fiber implants in metastatic spinal disease. The retrospective nature of the studies, along with confounding factors and selection bias, affects comparisons between standard titanium implants and the new carbon-fiber alternatives. Although some methodological approaches, such as pair-matching or stratification in the retrospective data, might mitigate certain confounding factors, the most valuable study design remains prospective randomized controlled trials, and so we are currently performing such a study.
From a clinical perspective, several topics need to be addressed regarding the materials used for internal fixation. The cost difference between standard titanium and carbon-fiber screws is substantial. Decreased levels of artifacts in radiological examinations, reduced dose scattering, and shorter planning durations are surrogate outcomes reported in the literature. However, it remains unclear whether the economic difference between these systems justifies their use in everyda
Comments (0)