
Remember me

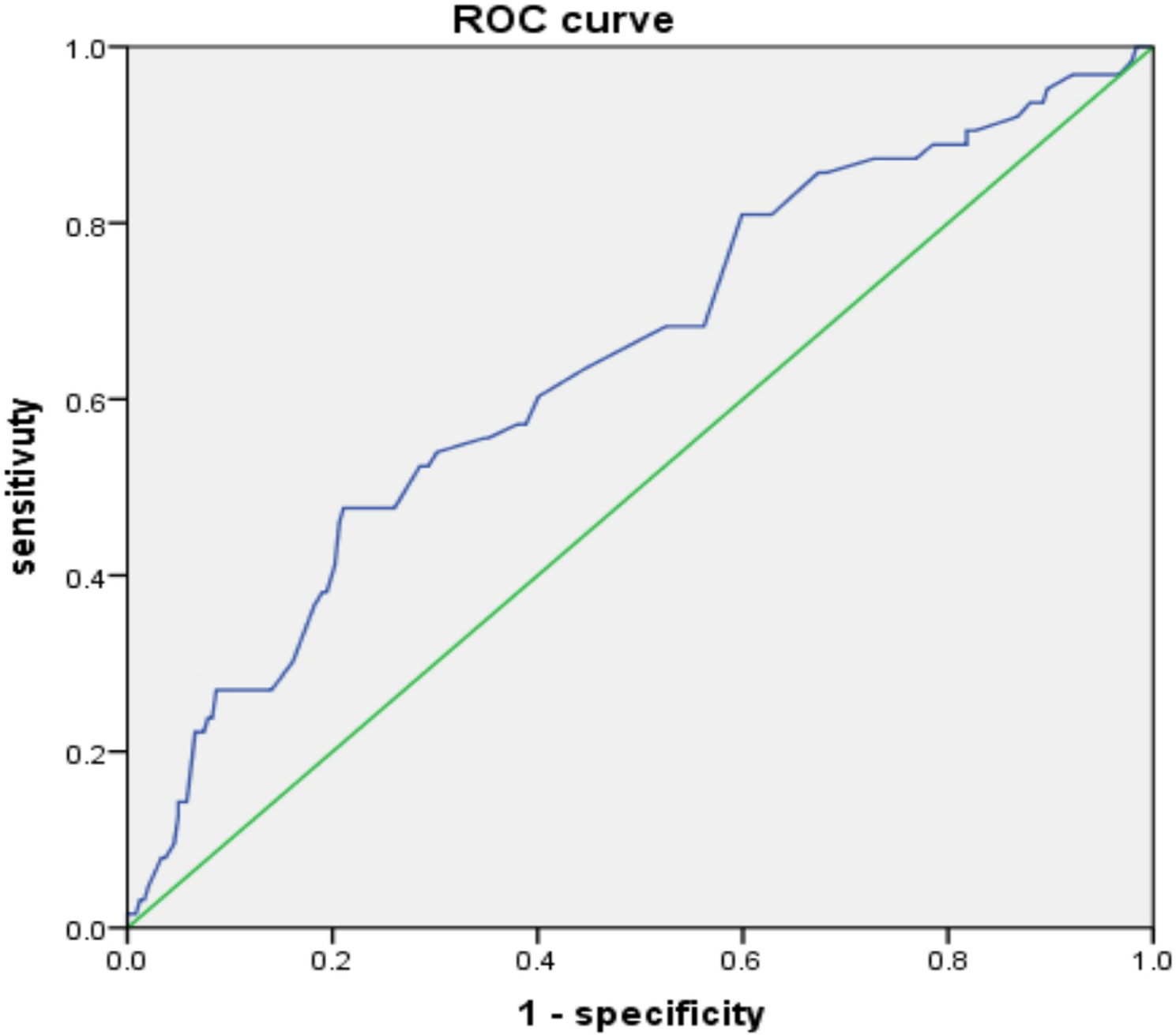
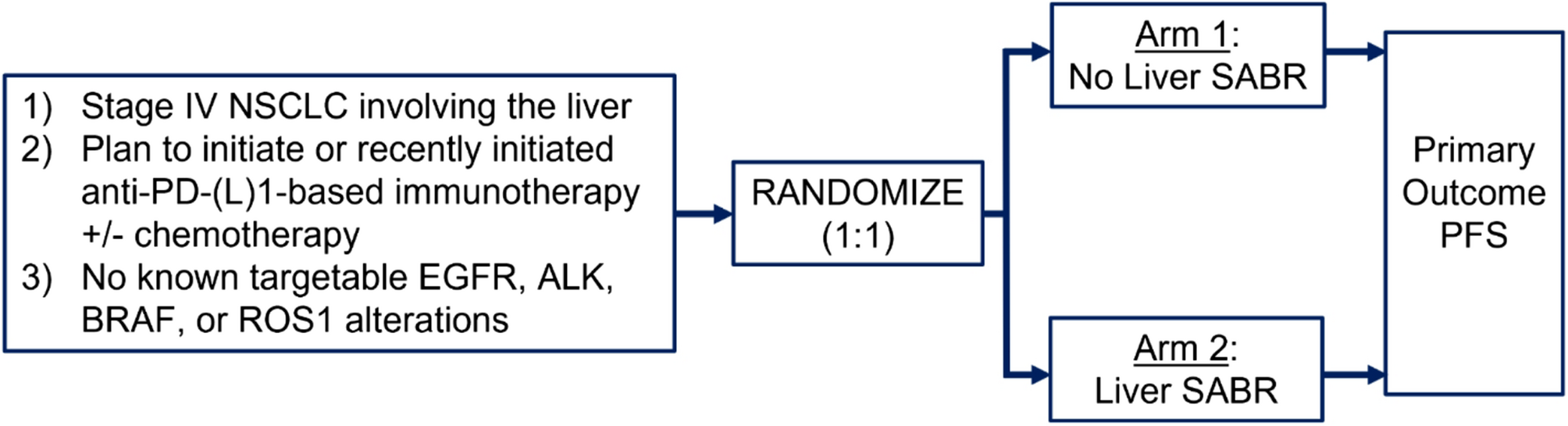
The primary objective of the HAMMER-NSCLC trial is to determine if adding liver SABR to standard-of-care first-line anti-PD-(L)1-based immunotherapy +/- platinum-based chemotherapy can improve median PFS in patients with metastatic NSCLC involving the liver. The secondary objectives include the safety and tolerability of adding liver SABR to anti-PD-(L)1-based immunotherapy regimens, overall survival, and hepatic progression. Correlative objectives include characterizing how liver SABR remodels both the liver tumor immune microenvironment and the systemic T cell response.
Study settingThe HAMMER-NSCLC trial is an investigator-initiated multicenter study sponsored by Memorial Sloan-Kettering Cancer Center (MSK) and run within the MSK Cancer Alliance in collaboration with the Miami Cancer Institute and Lehigh Valley Health Network. Patients will be accrued from all three participating institutions. The study, IRB 22–386, was approved by the institutional review board at MSK. Written informed consent is required for participation in the study.
Study designHAMMER-NSCLC is a multicenter, two-arm randomized phase II study investigating the addition of liver SABR to anti-PD-(L)1-based immunotherapy +/- platinum-based chemotherapy. The study schema is displayed in Fig. 1.
Fig. 1
HAMMER-NSCLC Trial Study Schema
Patient selection and eligibility criteriaPatients who are 18 years of age or older on the day of signing informed consent with histologically confirmed newly diagnosed NSCLC metastatic to the liver are eligible (Table 1). All NSCLC histologies are allowed. Patients are ineligible if their tumors have known targetable alterations in epidermal growth factor receptor (EGFR), BRAF, anaplastic lymphoma kinase (ALK), or c-ROS oncogene 1 (ROS1). Tumors with KRAS alterations are allowed. The patient must be able to receive liver SABR to all liver metastases; normal tissue constraints will be prioritized over target coverage. Patients must be planned for standard-of-care anti-PD-(L)1-based immunotherapy +/- platinum-based chemotherapy for at least three cycles. In addition to regimens containing a single anti-PD-(L)1 drug, immunotherapy regimens combining multiple immune checkpoint inhibitors, such as anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) immunotherapy with anti-PD-1 (e.g., ipilimumab plus nivolumab) or anti-PD-L1 (e.g., tremelimumab plus durvalumab) immunotherapy, are also allowed. Patients are ineligible if they have immunosuppression, Eastern Cooperative Oncology Group (ECOG) performance status ≥ 3, or significant baseline liver function test abnormalities (i.e., total bilirubin > 1.5x upper limit of normal [ULN] and/or AST/ALT > 5x ULN).
Table 1 Eligibility CriteriaPD-L1 TPS testingPD-L1 tumor proportion score (TPS) evaluation will follow institutional standards using validated antibodies such as E1L3N, SP263, and 22C3. PD-L1 TPS ≥ 50% is defined as membranous staining of any intensity in at least 50% of tumor cells. Central review is not mandated; however, PD-L1 scores and associated antibodies will be recorded systematically to ensure consistency.
Treatment planPatients will be randomized to standard-of-care treatment with anti-PD-(L)1-based immunotherapy +/- platinum-based chemotherapy (Arm 1) or standard-of-care treatment with anti-PD-(L)1-based immunotherapy +/- platinum-based chemotherapy plus liver SABR (Arm 2). Liver SABR will be delivered to all liver metastases and prescribed as per institutional standards in 3 to 6 fractions to 60 to 30 Gy (Table 2). Radiation dose and fractionation is determined by dose to the normal tissues including the liver (Table 3). Adherence to normal tissue constraints will be prioritized over target coverage. Patients should be preferentially planned with a Tier 1 dose and fractionation. If liver and/or organ at risk dose/volume constraints cannot be met with Tier 1 dose and fractionation schemas, Tier 2 schema should be considered. If not achievable with Tier 2 dose and fractionation schemas, then the Tier 3 schema should be considered. Dose-painting can be used as needed to maximize radiation dose to the tumor while limiting dose to adjacent healthy organs. If normal tissue constraints cannot be reasonably met with the Tier 3 dose and fractionation schema then the patient should be taken off study and proceed with standard of care therapy. For patients on the experimental Arm 2, it is preferred that liver SABR be completed before initiation of standard of care anti-PD-(L)1 therapy. Liver SABR must be completed prior to the third cycle of anti-PD-(L)1 or within 90 days of anti-PD-(L)1 therapy initiation, whichever is sooner. Liver SABR will be delivered in a week during which the patient receives no chemotherapy. Patients will thereafter continue anti-PD(L)−1-based immunotherapy +/- platinum based chemotherapy as per standard of care, as dictated by tumoral PD-L1 status. Liver SABR can be on the same week or even day as anti-PD-(L)1 and/or anti-CTLA-4 therapy.
Table 2 Dose constraints for target volumes and organs at riskTable 3 Suggested dose and fractionation schedulesAssessment and follow upThe radiation oncology service will conduct weekly interviews, physical examinations, and assessments for grade ≥ 3 toxicities during radiation treatment and again at 7 weeks post randomization (+/- 14 days) as part of the standard of care. Toxicities will be graded by Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. Adverse events will include grade ≥ 3 non-hematological toxicities or grade ≥ 4 hematologic toxicities attributed to liver SABR. All treatment evaluations will be performed per the standard of care (Table 4). After liver SABR, patients will undergo routine standard-of-care restaging with a PET-CT and/or CT imaging every 2 months until progression. Restaging imaging can be obtained at any time at the discretion of the clinical team as symptoms dictate. The study will use Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 for disease evaluation. Patients who present with tumor progression will be treated according to standard practice per institutional guidelines.
StatisticsPrimary objectiveThe primary objective in this trial is to compare the median PFS in patients with metastatic NSCLC undergoing treatment with standard-of-care first-line anti-PD-(L)1-based immunotherapy regimens +/- liver SABR. PFS is defined as the time from randomization to progression (local or distant) or death and will be summarized using the Kaplan-Meier method and compared by the stratified log rank test. Stratification of the log-rank test aligns the analysis with randomization and will use the same strata as in randomization: standard-of-care treatment plan (i.e., anti-PD-[L]1 therapy alone versus anti-PD-(L)1 therapy in combination with platinum-based chemotherapy versus anti-PD-(L)1 in combination with anti-CTLA-4) and burden of disease (i.e., oligometastatic [≤ 10 metastases] vs. polymetastatic [> 10 metastases) [12].
Sample size justificationThe sample size is justified with respect to the primary objective of comparing PFS between the randomized treatment arms. The null hypothesis of similar PFS between treatment arms will be tested. If the true median PFS times are 6 and 10.4 months in the control and experimental treatment arms, a sample of 68 patients achieves 80% power based on a one-sided alpha 0.10 level log-rank test, corresponding to HR of 0.6. The 6-month PFS in the control arm is based on KEYNOTE 189 (anti-PD-1 + chemotherapy for non-squamous NSCLC), which reported a median PFS of 6.1 months in patients with baseline liver metastases [13], and KEYNOTE 407 (anti-PD-1 + chemotherapy for squamous NSCLC), which reported a median PFS of 6.4 months [14]. These calculations are further based on enrollment over a 3-year period with 1 additional year of follow-up so that all patients will be followed for at least 1 year (and up to 4 years of total potential follow-up for the first enrolled patient). Based on the number of metastatic NSCLC patients treated at MSK and other participating institutions, we expect that the accrual rate will be approximately 23 patients per year. Thus, we anticipate approximately 3 years will be required for enrollment, followed by 1 year of additional follow-up for a total duration of 4 years.
Comments (0)