
Remember me
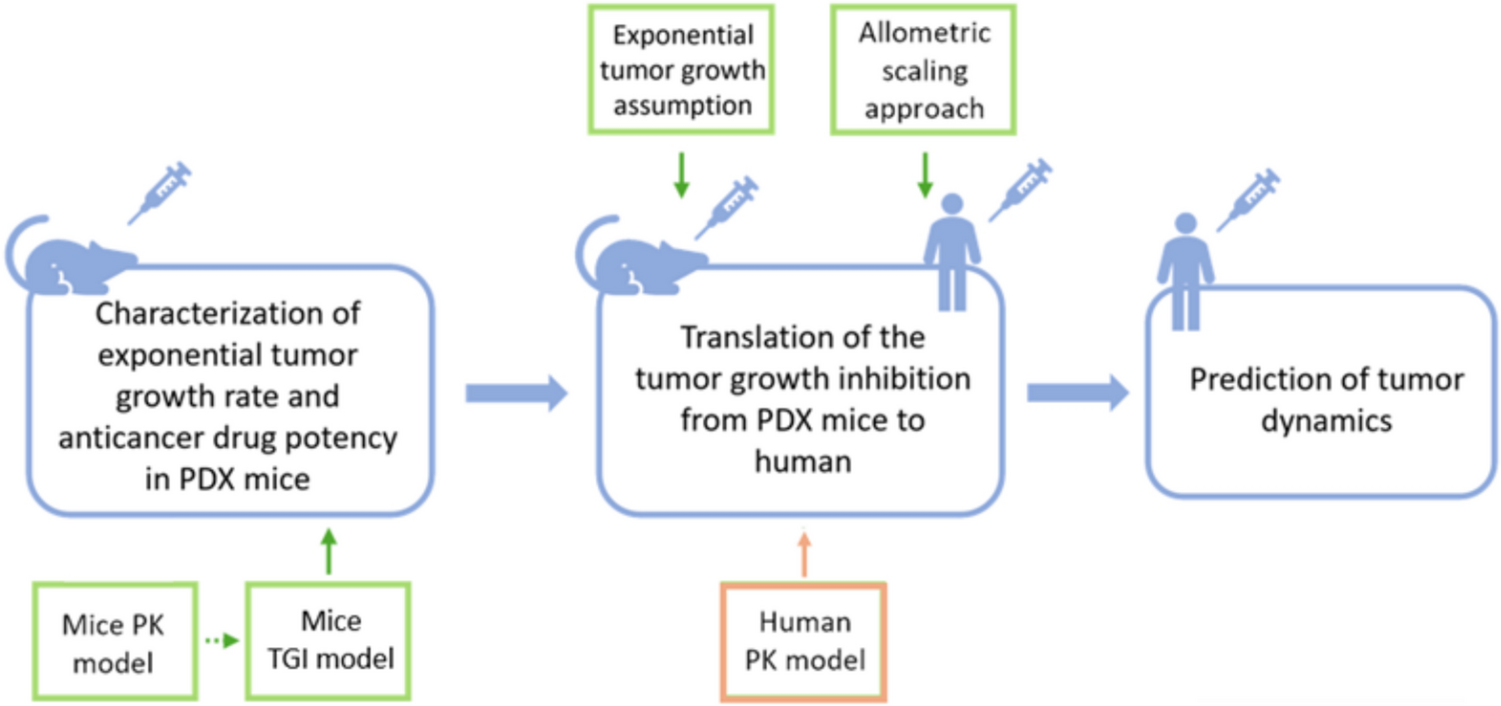
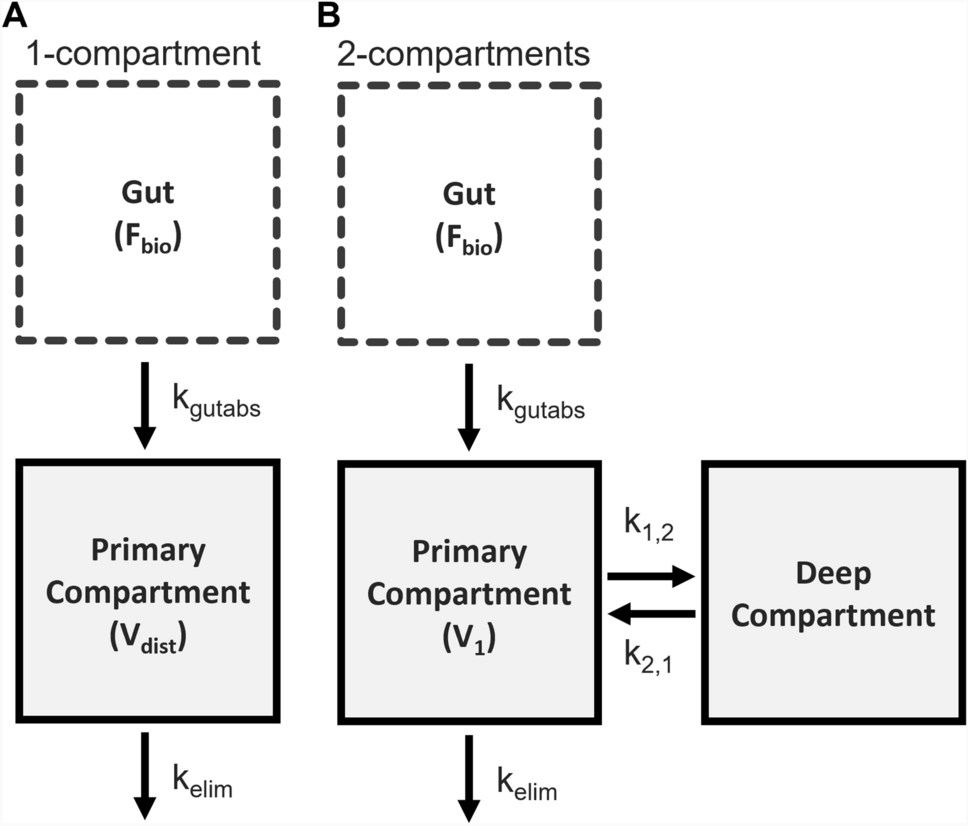
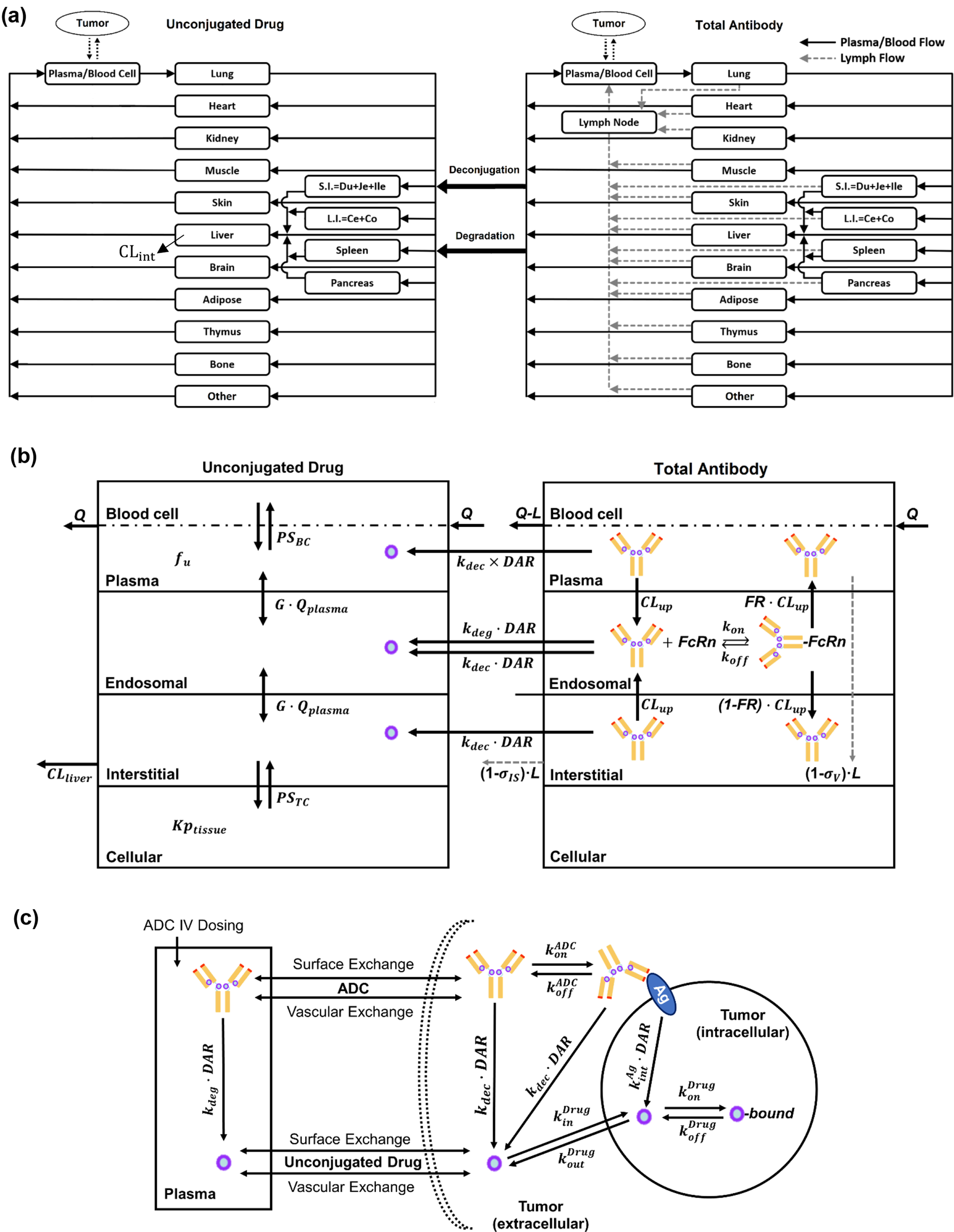

The model development process begins by defining a standardized structure, referred to here as the generic organ structure, as illustrated in Fig. 1. This structure is subsequently replicated to represent all major organs, culminating in the construction of the whole-body model depicted in Fig. 2. The generic organ structure is composed of four sub-compartments: vascular, endothelial, interstitial, and intracellular compartments. Some organs required additional organ-specific modifications which included adding an extra compartment to the kidneys that represented the lumped space of Bowman’s capsule, proximal tubules, and loop of Henle (BCPTLH), while the lung included an alveolar space to act as a potential drug administration/clearance site.
Fig. 1
Schematic presentation of the key pharmacokinetic processes and parameters of the generic organ model. The subscript \(}\) indicates an organ-specific parameter,\(Q\): blood flow,\(L\): lymph flow, \(JL\) and\(JS\): lymph flows through the large and small pores, respectively, \(PSL\) and\(PSS\): permeability surface area products of the large and small pores, respectively, \(\sigma L\) and\(\sigma S\): vascular reflection coefficients for the large and small pores, respectively,\(kin_\): internalisation rate constant of the conjugated drug-target complex,\(kup\): non-specific first-order uptake rate constant,, \(kde_\) and\(kde_\): vascular and intracellular degradation rate constants, respectively, and\(kred\): redistribution rate constant
Fig. 2
The full-PBPK model structure is defined in Simcyp Designer (Version 2, Certara UK, Sheffield, UK) [18]. Each greyish-blue colour represents an instance (copy) of the generic organ structure. Small red rectangles are assignment nodes that define the respective organ-specific parameters. Red solid and dashed blue arrows represent blood and lymph flow reactions, respectively. Solid black arrows represent other reactions. GIT: lumped gut compartment, ar: arterial compartment, ve: venous compartment, Ta: solid tumour compartment, tadln: solid tumour compartment draining lymph nodes, SC: subcutaneous administration site, scdln: subcutaneous draining lymph nodes, IM: intramuscular administration site, imdln: intramuscular draining lymph nodes
Soluble molecular species are exchanged between the vascular and interstitial compartments according to the two-pore model [19]. This model describes soluble species'diffusive and convective flux across the endothelial membrane’s small and large pores as a function of endothelial membrane porosity and species’ molecular size.
Cellular uptake of interstitial oligonucleotides was modelled to include two pathways. The first is a linear pathway representing the unsaturable non-specific uptake through fluid-phase endocytosis, adsorption to cell membrane and scavenger receptors [20,21,22], which applies to both conjugated and non-conjugated oligonucleotides. In parallel, the second, nonlinear pathway, which represents the specific saturable receptor-mediated endocytosis (RME) process, is active only for oligonucleotide conjugates. The latter involves concentration-dependent binding of the conjugate to a cell surface receptor and subsequent internalization of the complex, as observed for conjugated oligonucleotides [22, 23].
In the model, the receptors are modelled as being in continuous turnover, dynamically mediating ligand binding and dissociation whilst undergoing internalisation and recycling, as shown in Eq. 1 through Eq. 3. The internalisation step of the drug-target receptor complex is assumed to be followed by instantaneous dissociation of the bound ligand and recycling of the free receptor.
$$\begin\frac_\times _\right)}=\\ - \left(\left(ko_\times Dru_\times TARGE_-kof_\right.\right. \\ \left.\left. \times TARGET}_\right)\times _\right)+\left(ksy_ or}-kde_ or} \right. \\ \left. \times TARGE_\right)\times _+kre_\times TARGE_\times _\end$$
(1)
$$\begin\frac TARGET}_\times _\right)}\\ =\left(ko_\times Dru_\times TARGE_-kof_\right. \\ \left. \times TARGET}_\right)\times _-\left(kin_\right.\\ \left. \times TARGET}_\times _\right)\end$$
(2)
$$\frac_\times _\right)}=-\left(kde_\times TARGE_\times _\right)+kin_\times _\times _-\left(kre_\times TARGE_\times _\right)$$
(3)
where \(TARGE_\), and \(TARGE_\) are the target receptor concentration in the interstitial and intracellular spaces, respectively, \(_\), and \(_\) are the volumes of the interstitial and intracellular spaces, respectively, \(ko_\), and \(kof_\) are the binding and dissociation rate constants of the drug and the target receptor, \(Dru_\) is the total (protein bound and unbound) drug concentration in the interstitial space, \(Drug\_TARGE_\) is the drug-target complex concentration, \(ksy_ or}\) is the organ-specific zero-order synthesis rate of the target, \(kde_ or}\) is the organ-specific degradation rate constant of the target, \(kre_\) is the recycling rate constant of the intracellular target, and \(kin_\) is the internalisation rate constant for the drug-target complex.
Redistribution of oligonucleotide from the intracellular to the interstitial space was modelled through a first-order rate constant to account for the observed alignment between post-distribution plasma and tissue drug half-lives [24].
Oligonucleotide binding to plasma protein was modelled through the quasi-equilibrium approximation, i.e., assuming an instantaneous steady state between bound and free fractions. The free fraction in the tissue interstitial space is calculated using Eq. 4 and Eq. 5. Organ-specific albumin interstitial concentrations were obtained through running an albumin only simulation of the same model to steady-state. The alteration of molecule size due to binding to plasma proteins affects the calculated parameters in the two-pore model for tissue extravasation and renal filtration. On the other hand, it was assumed that protein binding does not impact the binding of conjugated oligonucleotides to target receptors, as these interactions are likely to occur at distinct sites [25].
$$K_=\frac_\times Al_}_+\epsilon }$$
(5)
where \(f_\) is the organ-specific fraction unbound of the drug in the interstitial space, \(K_\) is the equilibrium dissociation constant of albumin and the drug, \(Al_\) is the organ-specific albumin concentration in the interstitial space, \(f_\) is the fraction unbound of the drug in the vascular space, \(Al_\) is the albumin concentration in the vascular space, and \(\epsilon\) is a small positive constant (1E-6) to prevent division by zero when \(f_\) is one.
Equation 6 through Eq. 8 describe the drug kinetics in the generic organ’s compartments. The equations don’t include blood and lymph flow mediated transport of the drug in and out of the organ as these reactions are organ-specific and are added at the full-PBPK model assembly step discussed below.
$$\frac_\times _\right)}=-\left(kde_\times Dru_\times _\right)-\left(\left(\left(1-\sigma _\right)\times J_+\left(1-\sigma _\right)\times J_\right)\times _+\left(\frac_\times Pe_}_\right)-1}+\frac_\times Pe_}_\right)-1}\right)\times \left(f_\times Dru_-f_\times Dru_\right)+\left(\frac_\times Pe_}_\right)-1}+\frac_\times Pe_}_\right)-1}\right)\times \left(\left(1-f_\right)\times Dru_-\left(1-f_\right)\times Dru_\right)\right)$$
(6)
$$\frac_\times _\right)}=-\left(\left(ko_\times Dru_\times TARGE_-kof_\times _\right)\times _\right)-\left(ku_\times Dru_\times _-kre_\times Dru_\times _\right)+\left(\left(\left(1-\sigma _\right)\times J_+\left(1-\sigma _\right)\times J_\right)\times _+\left(\frac_\times Pe_}_\right)-1}+\frac_\times Pe_}_\right)-1}\right)\times \left(f_\times Dru_-f_\times Dru_\right)+\left(\frac_\times Pe_}_\right)-1}+\frac_\times Pe_}_\right)-1}\right)\times \left(\left(1-f_\right)\times Dru_-\left(1-f_\right)\times Dru_\right)\right)$$
(7)
$$\frac_\times _\right)}=kin_\times _\times _+ku_\times Dru_\times _-kre_\times Dru_\times _-\left(kde_\times Dru_\times _\right)$$
(8)
where \(PSS\) and \(PSL\) are the permeability surface area products for the small and large pores, respectively, \(PeS\) and \(PeL\), are the Peclet’s numbers for the small and large pores, respectively. PS and Pe calculation are described in detail elsewhere [26]. \(\sigma _\), and \(\sigma _\) are the vascular reflection coefficients for the small and large pores, respectively, and are calculated as the unbound-fraction-weighted average of the reflection coefficients for both bound and unbound drug as shown in Eq. 9 and Eq. 10.
$$_=_\times _+_\times (1-_)$$
(9)
$$_=_\times _+_\times (1-_)$$
(10)
Whole-body PBPK modelThe whole-body PBPK model used in this study was based on a published cross-species model [26] which has been adapted to fit the specific needs of this modelling effort. Briefly, the model included compartments that represent the lungs, lymph nodes, heart, gut, liver, spleen, pancreas, muscle, skin, bone, brain, thymus, adipose, and kidney, which correspond to 98% of the body weight. The rest of the body is modelled as “other” compartment to balance the total body weight and blood flow rates. In addition, the model was further adapted by separating small subcutaneous (SC) and intramuscular (IM) compartments as well as their draining lymph nodes from the total skin, muscle, and lymph node compartments, respectively. This allowed the mechanistic modelling of the absorption process following SC or IM administration. A general schematic of the adapted model structure is shown in Fig. 2.
PBPK model parametersPhysiological parameters for mice and rats were used as described previously [26]. This includes organ volumes, sub-compartmental volumes, lymph flows, blood flows, and organ-specific two-pore parameters. The two-pore parameters for the added SC compartment and its draining lymph nodes were assumed to be the same as the skin and lumped lymph nodes compartment, respectively. The sub-compartment volumes and the blood flow rate for the SC site are calculated based on the assumption that the SC dose is diluted in the total flow of the SC site’s watershed area before reaching the lymph nodes. Lymphatic watershed areas are distinctly defined areas of the skin that separate territories from each other and contain relatively few lymph collectors [27]. On average, a lymphatic watershed area constitutes 20% of the skin in mice and 6% in rats [28, 29]. The volume of lymph nodes draining the SC site and sub-compartmental volumes, are calculated under the assumption that the proportion of SC draining lymph node volume to the lumped lymph node compartment volume is consistent with the proportion of SC volume to the total body volume, i.e., SC site has the same lymph node density as the average across the whole body. All physiological parameters are provided in Supplement 1.
Data collection and modelling approachAll PK data (plasma and/or tissue concentration–time profiles) of conjugated and non-conjugated ASOs and sirRNAs in mice and rats used in this analysis was sourced from literature, as listed in Table 1.
Table 1 Summary of published data used to develop and validate the PBPK model. ASO: anti-sense oligonucleotides, IV: intravenous, SC: subcutaneous, siRNA: small interfering RNAA stepwise approach was adopted to the model parameterisation. First, a non-conjugated ASO data for the intravenous (IV) administration in rats reported by Zhang et al. [30] were used to estimate the linear uptake (\(ku_\)) and redistribution (\(kre_\)) rate constants for each of the observed tissues. For the unobserved tissues, the uptake rate constant was fixed at the macropinocytosis rate in FcRn-expressing endothelial cells of 0.9 h−1 [26], while redistribution was assumed to be zero. Those parameters were assumed to be tissue-specific and to be conserved for different oligonucleotide molecules and across species. The parameterised rat model was at first verified against independently measured rat ASO data [31,32,33]. In the next step, the cross-species aspect of the model was evaluated by applying it to the non-conjugated ASO data measured in mice [34,35,36]. The SC absorption model was verified using data from [37] which included liver concentration data after SC administration of a non-conjugated ASO in mice.
Parameter estimation was performed using a variant of the simulated annealing algorithm [40], a stochastic global optimisation method implemented in the stats package of R [41]. The goodness-of-fit was assessed by visual inspection of observation-overlaid predictions, residual plots, and the reduction of the objective function value.
In the second step, RME was added to the model to account for increased target-tissue uptake of conjugated oligonucleotides. The model was parameterised for the ASGPR-GalNAc receptor-ligand pair using literature values for the receptor abundance, turnover in mice, and ligand binding affinities as summarised in Table 2. The model was validated against IV- and SC-administered GalNAc-conjugated siRNA plasma and liver PK data from [38, 39]. No fitting of any system parameters was performed for the conjugated oligonucleotide data.
Table 2 Receptor-mediated endocytosis model parameters. *\(ksy_=kde_\times TARGE_\left(0\right)\)Oligonucleotide drugs are typically highly protein-bound [46]. However, specific values for most of the compounds used in this analysis have not been reported, and therefore, a fraction unbound in plasma of 0.15 was assumed. It was also assumed that nuclease clearance from plasma is negligible (\(kde_=0 h^\)) since all the examples used in this analysis had chemically modified nucleotides to confer nuclease stability. The main clearance mechanism from the systemic circulation is renal filtration which is included in the model as shown in (Eq. 11).
$$RF=GFR\times \left(_\times f_+_\times \left(1-f_\right)\right)\times Dru_$$
(11)
where \(\text\) is the rate of drug loss from renal vascular sub-compartment through renal filtration, \(GFR\) is the glomerular filtration rate, \(_\) and \(_\) are the glomerular sieving coefficients (GSC) for the free and bound drugs, respectively, \(f_\) is the fraction unbound of the drug in the vascular space, and \(Dru_\) is the drug concentration in the renal vascular sub-compartment.
The GSC is a factor that accounts for the size-dependent exclusion of soluble species from glomerular filtration and is calculated according to the empirically fitted formula in [26]. Intracellularly, oligonucleotides are cleared through a linear term (\(kde_\)) representing nuclease clearance which was allowed to vary for each specific sequence and chemical modification.
Model outputsAll model states were modelled as concentration species in molarity units. To facilitate comparison to observed data, whole organ concentrations in μg/g (or μg/mL for plasma) were computed as per Eq. 12 assuming a density of 1 g/mL for all organs for simplicity.
$$D_=\frac^_\times _}^_}\times _\times \frac^}_}$$
(12)
where \(D_\) is the observable drug concentration in the organ \( \text\) in μg/mL (plasma) or μg/g (other tissues), \(_\) is the volume of sub-compartment \(k\) of the organ \( \text\), \(Dru_\) is the drug concentration in the sub-compartment \(k\) of the organ \( \text\), \(_\) is the molecular weight of the drug, and \(_\) is the density of the organ \(or\).
Sensitivity and simulation analyses of targeted organ uptakeA local sensitivity analysis (LSA) was performed to evaluate the impact of individual model parameters on target organ uptake, quantified as the fraction of the administered dose internalized (\(_\)), as described by Eq. 13. The LSA was based on the PBPK model developed for the GalNAc-conjugated siRNA from the Nair et al. study [39] as a representative example. Both drug-specific parameters, such as the unbound fraction in plasma and intracellular degradation rate constant within the target organ; and system-specific parameters, including the internalization rate constant of the drug-receptor complex, dissociation rate constant, receptor recycling rate, uptake rate constant of the unobserved tissues, and receptor abundance in the target organ, were analysed. This allowed us to identify the most influential parameters governing drug uptake dynamics in the target organ.
$$_=\frac_^_\times _\times _ dDru_}$$
(13)
In addition, multiple model simulations were conducted to explore the role of receptor saturation on tissue-specific targeting, using the same example compound as in the LSA. The first set of simulations examined the relative contributions of receptor-mediated endocytosis (RME) and nonspecific uptake to the total amount of drug internalized by the target organ. To assess the dose-dependence of targeting specificity, we compared the fraction of the dose internalized by the target organ across a range of doses (0.1–50 mg/kg) for both conjugated and unconjugated compounds. Lastly, we explored the potential for enhancing targeting specificity through a sustained release formulation [47, 48]. This formulation assumed zero-order release over a given period for saturating doses of the conjugated compound, allowing us to investigate whether prolonged exposure could improve specificity for the target organ.
Modelling platformThe PBPK model was developed based on a biologics PBPK platform implemented in Simcyp Designer (Version 2; Certara UK, Sheffield, UK) [18]. The model building process starts with developing a generic structure of an organ as shown in Fig. 1. The whole-body PBPK model is then built automatically by adding intercompartmental flow reactions and two-pore exchange, while generating all required parameters and assignment rules. The automation allows large PBPK models with several hundreds of ODEs and thousands of reactions, parameters, and rules to be effortlessly and accurately generated without any human intervention required, in alignment with MIDD recommendations aimed at improving pharmacokinetic model reproducibility [49, 50]. More details on how the platform operates is provided in Supplement 2.
Comments (0)