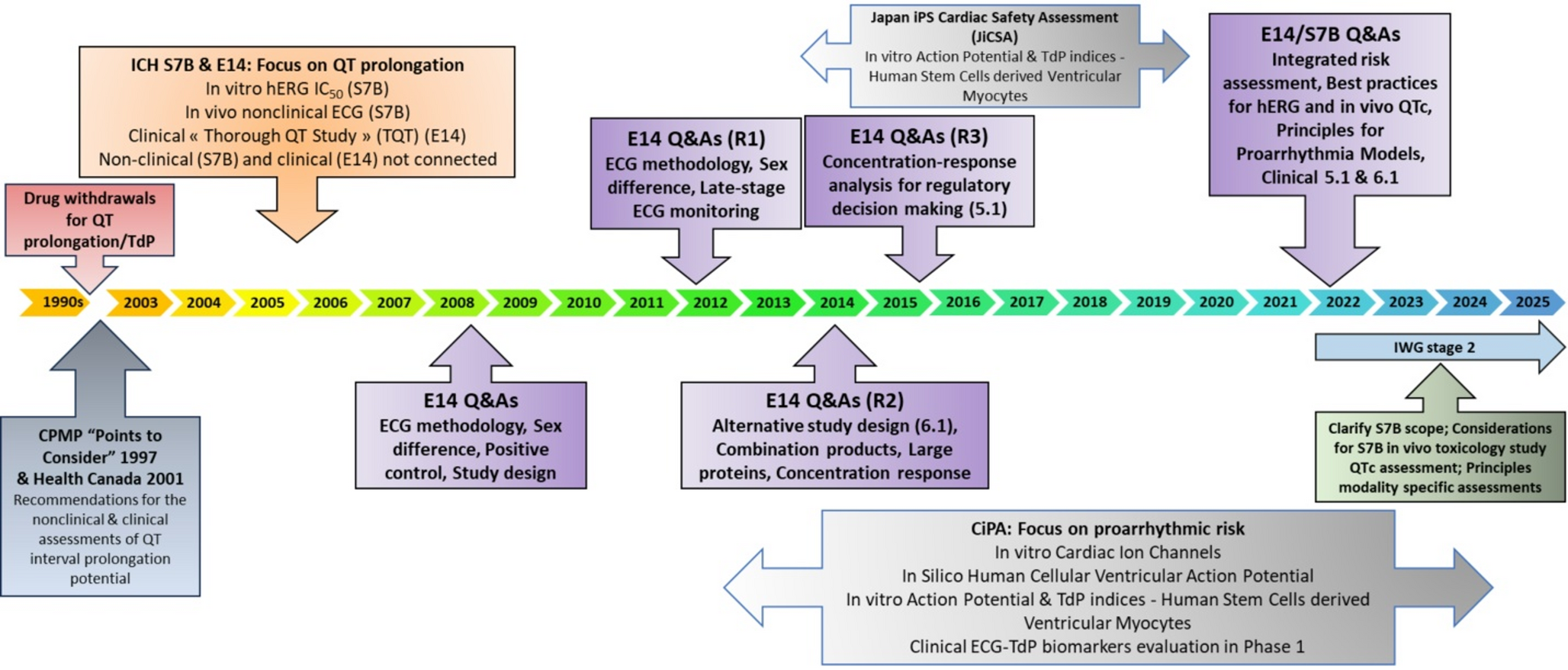
Over the past two decades, there have been significant changes in the way drug-induced QTc prolongation has been assessed, transforming both the clinical methodology and regulatory expectations. Drug-induced QTc prolongation, a measure of a drug’s potential to delay repolarization, has traditionally been a critical focus in the evaluation of new pharmaceuticals, especially small-molecule drugs with systemic bioavailability. Not all drugs, however, particularly those with low systemic exposure, require dedicated QT studies as per ICH E14 guidelines. For some drugs, especially those with known QTc effects, intensive ECG monitoring in later clinical phases may be preferred over a dedicated QT study. QT evaluations have expanded to include TQT studies, C-QTc analyses in early trials, and tailored QTc assessment based on drug-specific profiles (e.g., large targeted proteins) and therapeutic contexts. Meanwhile, pharmaceutical companies have improved at avoiding molecules likely to block the human ether-a-go-go-related (hERG) channel, the most common cause of QTc prolongation. However, there remain concerns within the scientific communities that intensified global focus on QTc safety may have unintended consequences such as the selection of suboptimal therapeutic agents, the abandonment of promising drug pathways, or the inadvertent oversight of other significant toxicities.
Initially, the TQT study was resource-intensive, incorporating multiple treatment arms, and requiring large sample sizes. A need clearly existed to reduce the sample size and make the study more efficient. In 2006, the FDA established the QT Interdisciplinary Review Team, which consisted of experts from the offices of Clinical Pharmacology, Biostatistics, and New Drug. Its primary function was to conduct comprehensive reviews of the nonclinical and clinical data packages related to QTc prolongation for most new drugs following the E14 guideline. This team ensured consistency in TQT study reviews and quickly recognized the importance of C-QTc analysis as a key method for interpreting TQT studies [2].
The C-QTc relationship, which examines the relation between drug concentration and QTc prolongation, has become increasingly prominent in QTc assessments. By utilizing C-QTc analysis, investigators can predict QTc effects at various clinically relevant concentration levels. Regulatory authorities globally implemented this approach in 2015, following the ICH’s release of E14 Q&A (R3) document endorsing C-QTc modeling as a primary analysis. This innovation led to two major outcomes in clinical development: sponsors could assess QTc effects during early-phase clinical trials, serving as a substitute for traditional TQT studies, and the TQT studies themselves became more efficient. The white paper on C-QTc modeling provided methodological recommendations, including study design, prespecified linear C-QTc model and reporting standards to support regulatory submissions [10]. Since its implementation, C-QTc modeling has reduced the number of participants required in TQT studies by up to 67%, depending on study design, while maintaining robust data on cardiac safety.
The 5.1 approach, which allows C-QTc modeling in early studies as a substitute for TQT studies, has been rapidly adopted by the pharmaceutical industry. The adoption of this approach has resulted in a notable reduction in TQT study submissions to the FDA, dropping from 75% in 2016 to 47% in 2017, with levels remaining steady in the following years. The observed decrease in TQT studies can be attributed to the increased use of both the 5.1 and 6.1 approaches. Notably, while the total number of submissions increased during this period, the volume of TQT-specific studies remained relatively stable. This shift reflects a broader trend towards more flexible and efficient assessment methods in drug development. One ongoing challenge, however, is achieving sufficient exposure levels in early-phase trials, a factor that has limited further reductions in the number of TQT studies.
In 2022, new Q&As for E14/S7B were implemented, allowing a ‘double-negative’ integrated nonclinical risk assessment as supplementary evidence, enabling 5.1 submissions to cover high clinical exposure rather than attaining the high-multiple of clinically relevant exposure requirement. This adjustment has the potential to further decrease the need for TQT studies. A ‘double-negative’ integrated nonclinical risk assessment includes: (1) hERG assays following best practices (S7B Q&A 2.1 [5]) showing low risk for parent and major human metabolites (S7B Q&A 1.1 [5]), demonstrated by hERG safety margins higher than a threshold defined based on the safety margins computed under the same experimental protocol for a series of drugs known to cause TdP; and (2) no evidence of QTc prolongation in an in vivo assay conducted according to ICH S7B at exposures covering high clinical exposures of parent and major human metabolites (S7B Q&A 3 [5]). When an integrated nonclinical risk assessment is used to support a drug with low proarrhythmic risk when evaluated in an alternative QT study, the in vivo assay must have sensitivity comparable to a TQT study (S7B Q&A 3 [5]). This can be accomplished by demonstrating that the minimal detectable difference (MDD) in the assay [11] is consistent with the reported MDD from a similarly designed assay (e.g., sample size, design, animal species, ECG methodology) that showed QTc prolongation with a positive control.
The QTc assessment for nirmatrelvir as described in Table 3 illustrates how a ‘double-negative’ integrated nonclinical risk assessment was used as supplementary evidence [12]. Nirmatrelvir was approved in 2023 in combination with ritonavir (PAXLOVID) for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults who are at high risk for progression to severe COVID-19, including hospitalization or death. The recommended dose is nirmatrelvir/ritonavir 300/100 mg BID with or without food for 5 days [13]. The QTc assessment focused on nirmatrelvir because the QTc effects of ritonavir 100 mg BID have already been characterized [14]. High fat meals increase nirmatrelvir Cmax by 1.6-fold [13]. Increased exposure was observed in patients with severe renal impairment compared to healthy subjects (Cmax: ~ 1.5-fold) [13]. In the review of the QTc assessment, the high clinical exposure scenario was considered as 150 mg/100 mg (nirmatrelvir/ritonavir) in patients with severe renal impairment under fed conditions. The exposure coverage for nirmatrelvir in the QTc assessment was only 1.5-times the high clinical exposure scenario [12]. This is less than the exposure coverage necessary to waive the requirement for a separate positive control. However, the applicant included a ‘double-negative’ integrated nonclinical risk assessment as supplementary evidence. The clinical and nonclinical data therefore supported concluding an absence of QTc prolongation like a TQT study. Examples of approved drugs that used an integrated nonclinical risk assessment under the 5.1 approach are QALSODY (tofersen) [15] and WAINUA (eplontersen) [16]. So far, no drug has successfully had an integrated nonclinical assessment to support low proarrhythmic risk under the 6.1 approach.
Table 3 Example of double negative integrated nonclinical data that provided supplementary evidence to support the lack of QTc prolongation in a small phase 1 study of nirmaterelvir/ritonavir [12]The white paper C-QTc model is a prespecified linear mixed-effect model that is used primarily to exclude a 10-ms mean increase in ∆∆QTc as measured by the upper bound of the one-side 95% confidence interval [10]. This simple model is recommended because it can be used for most study designs supporting QTc assessments, such as single ascending dose (SAD), multiple-ascending dose (MAD), and TQT studies. The linear model has been shown to have robust operating characteristics in simulation studies [17] and in a prospective, proof-of-concept study of five QTc prolonging drugs [18]. In our experience, for 70 drugs that were found to prolong the QTc interval, the linear white paper model was used for 60% of the reports to characterize the QTc effects.
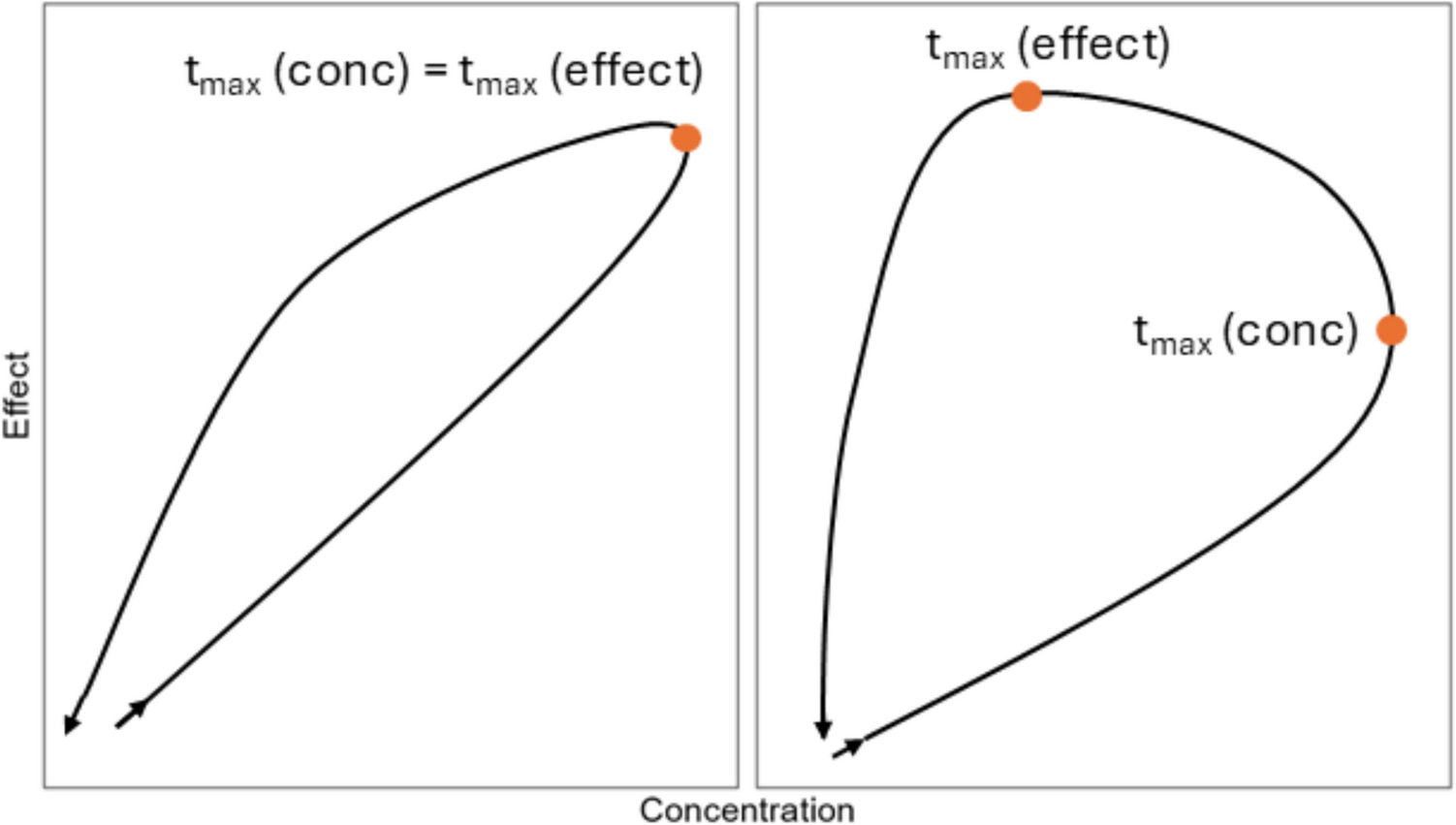
Mechanistic insights into QTc prolongation have also shaped these evolving methods. For instance, drugs that inhibit the hERG channel generally show a linear C-QTc relationship, whereas a nonlinear C-QTc relationship often indicates a different underlying mechanism or inhibition of multiple cardiac ion channels. Caution in using nonlinear models without a solid mechanistic rationale was highlighted in the case of macimorelin [19].
The macimorelin case illustrates the challenges with predicting the QTc changes at doses not studied when the mechanism for QTc prolongation is unknown [19]. The TQT study was a 3-way cross-over study with a single supratherapeutic dose. An increase in the QTc interval was observed following a single dose; however, the increase in QTc was delayed relative to macimorelin concentrations. The mechanism for the observed delay was unknown. An effect-compartment could be used as a model for such data, which would suggest less QTc prolongation at lower doses. While reviewing the TQT study, the FDA reviewer evaluated a SAD study of macimorelin as well, in which serial ECG/PK data were collected at 3 dose levels, with the highest dose being the same as the single dose from the TQT study. A similar delay and magnitude of QTc prolongation were observed for the overlapping dose (Table 4). However, the time-course for the QTc interval for the two lower doses were identical to that of the high dose. This observation therefore challenges the use of a simple effect-compartment model to describe observed data. Relying on a C-QTc model based solely on the TQT study using a single supratherapeutic dose of macimorelin might have underestimated the QTc effect, underscoring the importance of dose response data in the QTc assessment if the mechanism is not mediated by hERG inhibition.
Table 4 Example of QTc prolongation with unknown mechanism [19]Challenges with using simple C-QTc models have also been observed for other QTc prolongers such as oliceridine [20] and gepirione [21]. In both cases, apparent time-dependent QTc prolongation effect was observed. In the case of oliceridine, QTc prolongation decreased after repeat dosing due to an unknown mechanism. Two TQT studies were conducted. In the single-dose TQT study where therapeutic (3 mg IV infusion) and supratherapeutic (6 mg IV infusion) doses were tested, dose-dependent QTc prolongation (3 mg: 7 ms [upper two-sided 90% CI: 9 ms]; 6 mg: 12 ms [14 ms]), which occurred after peak oliceridine plasma concentration, was observed [22]. In the multiple-dose TQT study where intermittent dosing over 24 h to the maximum daily cumulative dose of 27 mg (maximum daily dose recommended by the label) was evaluated, QTc prolongation, which peaked at 9 h post the 1 st dose (mean QTcF 11 ms [upper two-sided 90% CI: 13 ms]), diminished after 12 h [22]. In the case of gepirone, C-QTc analysis revealed different relationships on Day 1 (non-linear) and Day 7 (linear) with less QTc prolongation on Day 7 despite higher plasma concentration. The mechanism is unknown. These challenges create opportunities for further advancement of C-QTc modeling, as supported by mechanistic data.
Various models, including in silico, in vitro, ex vivo and in vivo models, can be used as part of a comprehensive risk assessment strategy to evaluate the proarrhythmic risk of QTc-prolonging pharmaceuticals in humans. The utilization of in vitro and in silico models aligns with the 3R (reduce/refine/replace) principles, effectively reducing animal use in research. For these proarrhythmia risk prediction models to be considered valid for regulatory purposes, they must follow general principles. These include having a clearly defined endpoint consistent with the context of use, a specified domain of applicability/scope with acknowledged limitations, a mechanistic interpretation of the model, and the ability to propagate uncertainty from model input to model prediction (as outlined in S7B Q&A 4.1 [5]). These principles align with general principles of ICH M15 for model-informed drug development [23]. The implementation of these models could potentially address practical limitations in evaluating supratherapeutic doses in healthy subjects or patients and provide valuable insight into the clinical implications of small changes in QTc interval, particularly in the context of drug-drug interactions.


Comments (0)