Description of Bioanalytical MethodsAssay for Detection of Anti-tocilizumab Antibodies
Both the phase I and the phase III studies utilized an electrochemiluminescence (ECL) assay bridging format with labeled BAT1806. Serum samples were pre-treated in an acid dissociation step to ensure suitably sensitive detection of ADA in the presence of on-board drug levels.
The ADA assay was validated in accordance with global regulatory recommendations [2, 3, 11]. Per current recommendations, a single ADA assay using BAT1806 as the antigen was applied for detection of ADA in samples collected from participants treated with either BAT1806 or TCZ sourced from the EU (TCZ EU) or sourced from the US (TCZ US) [12]. Use of a single ADA assay was justified by the demonstration of antigenic equivalence, i.e., equivalent reactivity of anti-BAT1806-positive control antibody with TCZ EU and TCZ US (Supplementary Fig. S1a, b). In addition, it was shown that exchanging BAT1806 and TCZ as capture and detection agents did not result in differences in detecting various concentrations of positive control ADA.
Screening assay sensitivity was 7 ng positive control antibody/ml in the absence of TCZ and tolerant to 200 µg TCZ/ml for the detection of ≥ 10 ng (BAT1806-002-CR study) or ≥ 16 ng (BAT1806-001-CR study) positive control antibody/ml. While concentrations of the target protein, IL-6R, of up to 600 ng/ml did not interfere with detection of the positive control signal, false-positive signals were detected in the screening assay used for the BAT1806-002-CR study when the IL-6R concentration was higher than 150 ng/ml; however, the impact of soluble IL-6R interference was not determined for the confirmatory assay.
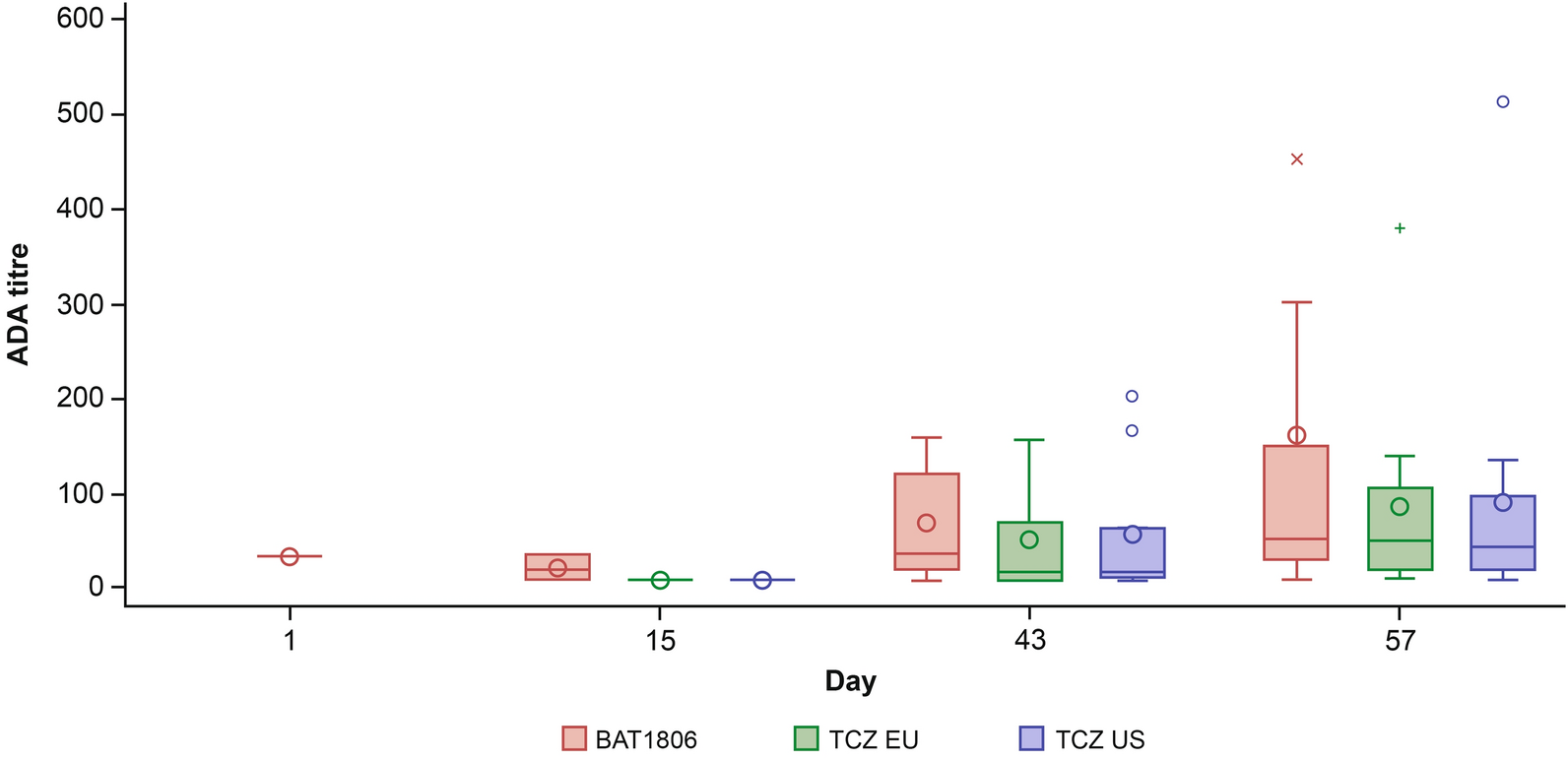
ADA positivity was based on the confirmatory ADA assay demonstrating inhibition in the presence of excess TCZ (5 μg BAT1806 per ml). Confirmed ADA-positive samples were further characterized for the magnitude of the signal (titer) in the ADA assay format. In the BAT1806-002-CR study, ADA titer was defined as the highest sample dilution yielding a signal greater than or equal to the assay cut point (0.1% false-positive rate) multiplied by the minimum required dilution ([MRD] = 20). In the BAT1806-001-CR study, ADA titers were presented as interpolated values between the highest dilution that was above the titration cut point and the adjacent dilution below the titration cut point, multiplied by the MRD of 10.
Cut points were calculated by statistical analysis [13] of signals for 54 individual sera from treatment-naive healthy donors for the BAT1806-001-CR study and using pre-treatment samples from 51 patients with RA, the study population for the BAT1806-002-CR study. The cut points were based on false-positive detection rates of 5%, 1%, and 0.1% for the screening, confirmatory and titration tiers, respectively.
NAb Assay to Characterize the Neutralizing Potential of Anti-BAT1806 and Anti-TCZ Antibodies
A competitive ligand-binding ECL assay was used to measure capacity of NAb to inhibit the binding of labeled BAT1806 to recombinant human IL-6R coated on microtiter plates. The NAb assay sensitivity was 138 ng positive control antibody/ml and drug tolerance was 25 µg drug/ml for detection of 500 ng positive control antibody/ml using a cut point corresponding to a 1% false-positive rate. The assay demonstrated that sIL-6R up to 100 ng/ml did not interfere with detecting the positive control antibody at a concentration of 100 ng/ml or higher. Adequate sensitivity and drug tolerance of the NAb assay were confirmed by the high concordance of the ADA and NAb assay results: 95% of confirmed ADA-positive samples from BAT1806-002-CR were also NAb-positive.
Assay for Quantitation of BAT1806 and TCZ (EU and US) in Human Serum
In both studies, serum concentrations of BAT1806 and TCZ were quantified using a ligand-binding enzyme-linked immunosorbent assay where BAT1806 or TCZ in human serum were bound to recombinant human IL-6R coated on 96-well microtiter plate wells, followed by detection of bound drug using a horseradish peroxidase-conjugated anti-tocilizumab non-paratope-specific, anti-idiotype monoclonal antibody (Bio-Rad Cat. No. HCA257). The validated assay range was 0.2–10.0 μg/ml.
Assessment of Clinical Immunogenicity
The immunogenicity analyses described here were based on post hoc analyses of participant-level data for the Safety Analysis populations in the BAT1806-002-CR and BAT1806-001-CR studies.
The BAT1806-001-CR study [10] evaluated comparative PK following a single intravenous (IV) administration of 4 mg/kg of BAT1806, TCZ EU, and TCZ US to 129 healthy Chinese male volunteers in a randomized, double-blind, 3-arm, parallel-group design. The primary objective of the study was to establish a pairwise PK biosimilarity between BAT1806 versus TCZ EU, BAT1806 versus TCZ US and TCZ EU versus TCZ US. Participants were randomized in a 1:1:1 ratio and followed up for 57 days. BAT1806-002-CR [8, 9] was a 52-week, randomized, double-blind, parallel-group study to compare the efficacy and safety of BAT1806 to TCZ EU in 621 patients with RA with an inadequate response to methotrexate (MTX). The primary objective of this study was to demonstrate the equivalent efficacy of BAT1806 and TCZ EU in participants with RA inadequately controlled by MTX. This study comprised three periods: Treatment Period 1 (TP1) (weeks 0–24); Treatment Period 2 (TP2) (weeks 24–48); and a 4-week safety follow-up period. Eligible patients were randomized in a 2:1:1 ratio to one of three treatment groups BAT1806 (TP1)/BAT1806 (TP2), TCZ (TP1)/TCZ (TP2) or TCZ (TP1) followed by BAT1806 (TP2) each to receive intravenously administered tocilizumab every 4 weeks at a dose of 8 mg/kg. Prior treatment with more than two biological disease-modifying anti-rheumatic drugs (DMARDs), or an IL-6 inhibitor or any targeted synthetic DMARD was not permitted. The patients with RA treated in the BAT1806-002-CR study received concomitant MTX (stable dose level of 10–25 mg MTX/week). Participants were expected to remain on their stable dose of MTX throughout the study.
Key characteristics of each study, as well as the variables used in the immunogenicity analyses, are presented in Table 1. ADA evaluable participants were defined as those who had an ADA result at baseline (either ADA-positive or ADA-negative) and had at least one post-treatment result. A participant was considered ADA/NAb-positive if they had at least one ADA/NAb-positive sample during the period (excluding baseline/week 0) up to and including the end of the treatment period. ADA responses were defined as follows: ‘Pre-existing ADA’ (antibodies reactive with the biologic drug that were present in participants before study treatment) and ‘Treatment-induced ADA’ (ADA developed de novo [seroconversion] following biologic drug administration [i.e., formation of ADA any time after the initial drug administration in a participant without pre-existing ADA]) [14]. Treatment-induced ADA were further subcategorized as either ‘Transient’ or ‘Persistent’. in line with recommendations from Shankar et al. [14] (Supplementary Material 1). No data imputation for missing ADA data was applied and data were presented ‘as observed’. To determine the relative impact on efficacy, American College of Rheumatology (ACR) 20, ACR50 and ACR70 response rates, as well as Disease Activity Score-28 for rheumatoid arthritis with erythrocyte sedimentation rate (DAS28-ESR) were evaluated. Efficacy outcomes were analyzed on the full analysis set, which included all randomized participants. Safety was evaluated with respect to (i) treatment-related serious AEs by ADA status, and (ii) AEs corresponding to Preferred Terms within the broad Standardised Medical Dictionary for Regulatory Authorities query (SMQ) of ‘hypersensitivity’ by ADA status [15]. The safety analyses were performed on the safety analysis set, which included all randomized participants who received any treatment with the study drug.
Statistical Analysis
ACR20/50/70 responses and DAS28-ESR were descriptively analyzed in the full analysis set and for predefined subgroups (ADA/NAb status [in TP1 and TP2 separately and combined], region [Central Europe/Asia Pacific]), and treatment period. In BAT1806-002-CR, TP1 and TP1 and TP2 (combined), neither missing values nor intercurrent events were considered in the descriptive analysis.
The estimated response probability/change in DAS28-ESR scores from baseline for each treatment group with the 90% respective 95% confidence intervals for the between-treatment-group difference was calculated.
Product Quality
The analytical and biological characteristics of BAT1806 were assessed using comprehensive analytical techniques and in vitro assays to determine similarity with TCZ EU, TCZ US, and China sourced tocilizumab. The results of these analyses are presented elsewhere [16]. HMW content was determined by size exclusion-high-performance liquid chromatography (HPLC). The degradation trends of BAT1806 and TCZ for HMW-variant content were also compared under accelerated conditions (products stored at 25°C for 12 weeks).
Ethical Approval
BAT1806-002-CR was conducted in accordance with the Declaration of Helsinki and/or all relevant local regulations, in compliance with the International Council for Harmonisation Good Clinical Practice guidelines and according to the appropriate regulatory requirements in the countries where the study was conducted. BAT1806-001-CR was approved by The First Bethune Hospital of Jilin University, Changchun, Jilin, China. The patients/participants provided their written informed consent to participate in this study. Patient consent was not applicable for this comparison article.

Comments (0)