
Remember me
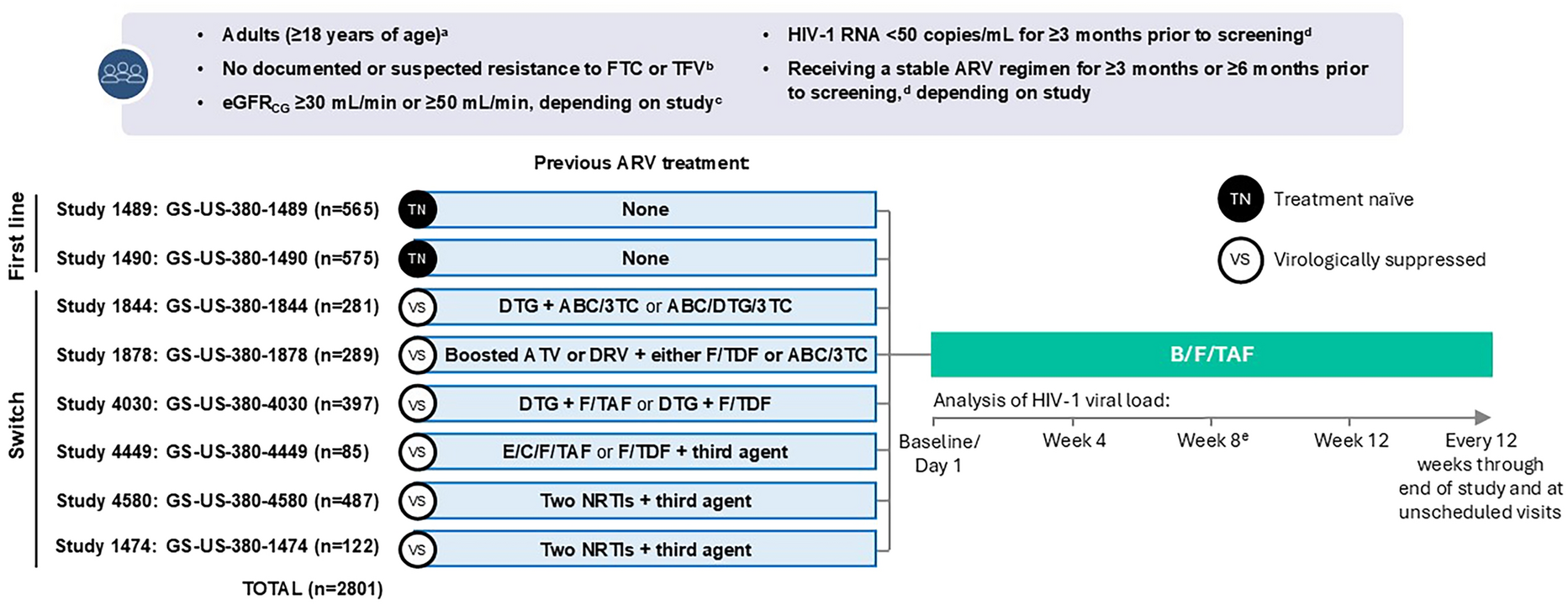
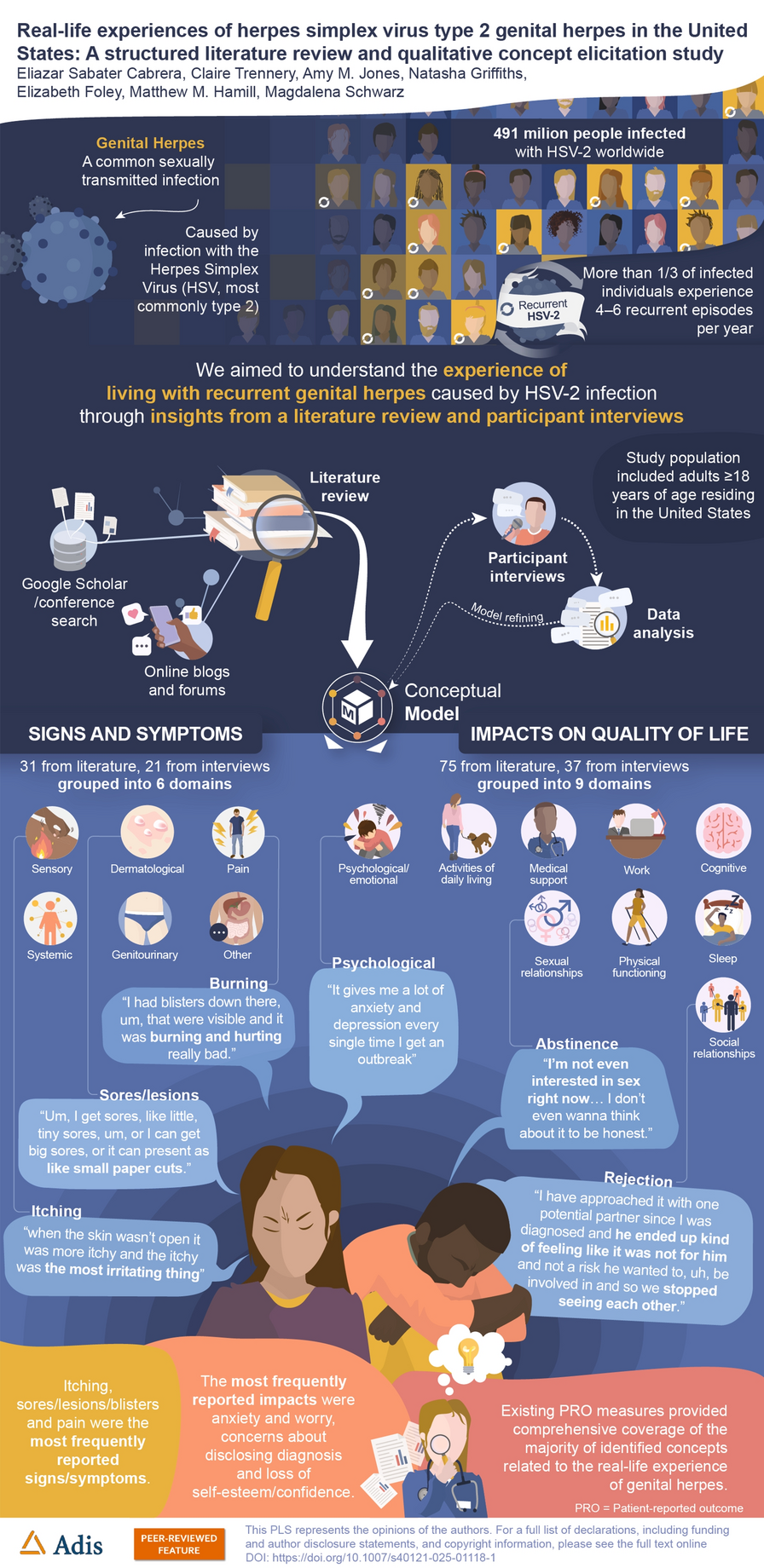
This randomized, double-blind, placebo-controlled, three-part, phase 1, FTIH, single and multiple dose-escalation study (ClinicalTrials.gov identifier, NCT05163522) was designed to assess the PK, safety, and tolerability of VH-280 administered after a standardized, moderate-fat, moderate-calorie meal (~600 cal with 30% from fat) in adults without HIV (Fig. 1). The study was conducted at a single inpatient clinical trial center in the USA between 10 January 2022 and 23 June 2023.
Fig. 1
FTIH study design. aFTIH first-time-in-human, MDZ midazolam, PiB powder-in-bottle, VH-280 VH4004280. aN refers to planned sample size. bWith the exception of the starting dose for cohort 1 in part 1, all doses were determined by a safety and dose escalation committee after review of pharmacokinetic and safety data
Adults aged 18–55 years (inclusive) with body weight between 50 and 100 kg (inclusive) and body mass index between 19 and 30 kg/m2 (inclusive) who were determined to be overtly healthy on the basis of medical evaluation, including medical history, physical examination, laboratory tests, and cardiac monitoring, were eligible to participate. Key exclusion criteria included alanine aminotransferase (ALT) > 1.5 times the upper limit of normal (ULN; single repeat test allowed during the screening period to determine eligibility); total bilirubin > 1.5 times ULN (isolated total bilirubin > 1.5 times ULN was acceptable if direct bilirubin < 35%); positive laboratory tests for hepatitis B, hepatitis C, HIV-1 antibody, or SARS-CoV-2; current or chronic history of liver disease; known hepatic or biliary abnormalities; history or presence of any conditions that could affect the absorption, distribution, metabolism, or excretion of the investigational drug; or sensitivity to any study medication.
Parts 1 and 2 were randomized using RandAll NG, a GSK-validated randomization software, and double-blinded, with participants and site staff blinded. The sponsor was blinded except for the statistician and clinical pharmacologist who had access to unblinded data for dose escalation decisions. Part 3 was open-label, and all participants received VH-280.
A safety and dose escalation committee, which included both sponsor and investigator/site staff, determined all doses, except for the starting single dose of 10 mg in part 1, after review of emerging PK and safety data. All visits for dosing and 7-day post–last-dose follow-up took place in the inpatient clinical trial center; outpatient visits were weekly, with the final outpatient follow-up visit occurring ~49 days after the last dose.
This study was conducted in compliance with legal and regulatory requirements and followed the ethical principles described in the Declaration of Helsinki. Advarra (Columbia, MD) reviewed and approved the study protocol, and all participants provided written informed consent before study initiation.
ProceduresSingle Ascending DosesIn part 1, single ascending doses of oral VH-280 powder-in-bottle (PiB) formulation were evaluated in five sequential cohorts. Participants were randomized (6:2) to receive either a single dose of VH-280 (10–900 mg) or placebo in each cohort. Two participants per cohort served as sentinel participants; 1 received blinded VH-280 and the other received blinded placebo. After review of 24-h safety data (e.g., vital signs, electrocardiograms, laboratory tests, and adverse events [AEs]), remaining participants received either VH-280 or placebo.
Plasma samples were collected from participants in all cohorts to characterize VH-280 PK, and urine and plasma samples were collected from participants in cohort 5 to characterize VH-280 metabolites.
Multiple Ascending DosesIn part 2, multiple ascending doses of once-daily oral VH-280 PiB formulation were evaluated in three sequential cohorts. In cohorts 1 and 3, participants were randomized (6:2) to receive either once-daily doses of VH-280 (100 or 350 mg, respectively) or placebo on days 1 through 14. In cohort 2, midazolam (MDZ) was used as a probe substrate to assess the potential of VH-280 to inhibit or induce cytochrome P450 3A (CYP3A) enzymes. In addition, coproporphyrin-1 (CP-1) was used as an endogenous biomarker probe [29] to evaluate the potential of VH-280 to inhibit organic anion transporter 1B1/1B3 (OATP1B1/1B3). In cohort 2, participants were randomized (8:2) to receive a single dose of MDZ 5 mg on days 1, 2, and 15, and VH-280 250 mg or placebo once daily on days 2 through 15. Samples of CP-1 were collected before (day 1) and after (day 15) multiple-dose administration of VH-280 250 mg to evaluate any impact of VH-280 on OATP1B1/1B3 inhibition.
Plasma samples were collected from participants in all cohorts to characterize VH-280 PK and from participants in cohort 3 to characterize VH-280 metabolites. Plasma samples were collected from participants in cohort 2 to characterize MDZ and 1-hydroxymidazolam PK and to measure CP-1. Urine and duodenal bile samples (using EnteroTracker® [EnteroTrack™, Aurora, CO]) were collected from participants in cohort 3 to characterize VH-280 metabolites.
VH-280 Tablet FormulationIn part 3, a single dose of VH-280 tablet formulation was evaluated in one cohort; all participants received VH-280 tablets at a dose of 450 mg. Data for the PiB and tablet formulations were compared.
Analytical MethodsValidated liquid chromatography–tandem mass spectrometry methods were used to analyze plasma for VH-280, MDZ, and 1-hydroxymidazolam PK and CP-1 biomarker assessments. Liquid chromatography–multiple-stage mass spectrometry, fluorine-19 nuclear magnetic resonance spectroscopy, and comparison of chromatographic retention times and mass spectrometry fragmentation patterns with authentic reference standards, as appropriate, were used to identify VH-280 metabolites in plasma, urine, and duodenal bile.
PK and Safety EvaluationsPharmacokinetic parameters evaluated included area under the plasma concentration–time curve from time 0 to infinity (AUC0–∞), AUC from time 0 to last quantifiable time point (AUC0–t), AUC from time 0 to 24 h (AUC0–24), maximum observed plasma concentration (Cmax), time to Cmax (tmax), and terminal half-life (t1/2). Safety assessments included AE incidence and severity; AEs leading to discontinuation of drug or study; vital signs and electrocardiogram changes; and maximum toxicity grade increase from baseline for laboratory hematology parameters, liver panel parameters (total bilirubin, direct bilirubin, alkaline phosphatase, ALT, and aspartate aminotransferase [AST]), total bile acids, lipid parameters (total cholesterol [TC] and low-density-lipoprotein cholesterol [LDL-C]), and creatine kinase. Adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA®) [30] and graded using the Division of AIDS criteria (version 2.1) [31].
Statistical AnalysesThere were no formal statistical hypotheses tested. For all study parts, sample size was based on feasibility, and no formal calculations were performed for power or sample size. For active dosing, a sample size of approximately six to eight participants was determined to provide PK estimates with acceptable precision and high probability to detect frequent AEs.
Pharmacokinetic parameters were calculated using standard non-compartmental methods with Phoenix WinNonlin 8.1 or higher (Certara, Radnor, PA), and PK and safety data were summarized descriptively using SAS® version 9.4 (SAS Institute Inc, Cary, NC). A mixed-effects linear model with fixed-effects terms for visit and random effects for participants was used for the comparison of MDZ, 1-hydroxymidazolam, and CP-1 PK parameters on days 2 and/or 15 versus day 1. A fixed-effects analysis of variance model fitting for treatment was used for the comparison of VH-280 tablet versus PiB formulation PK parameters. Power and analysis of variance models were used to assess dose proportionality of single and multiple ascending doses of VH-280. A mixed-effects analysis of variance model with a random intercept (for each participant) and fixed effect for day was used to estimate accumulation ratios after multiple daily doses of VH-280.
Comments (0)