
Remember me
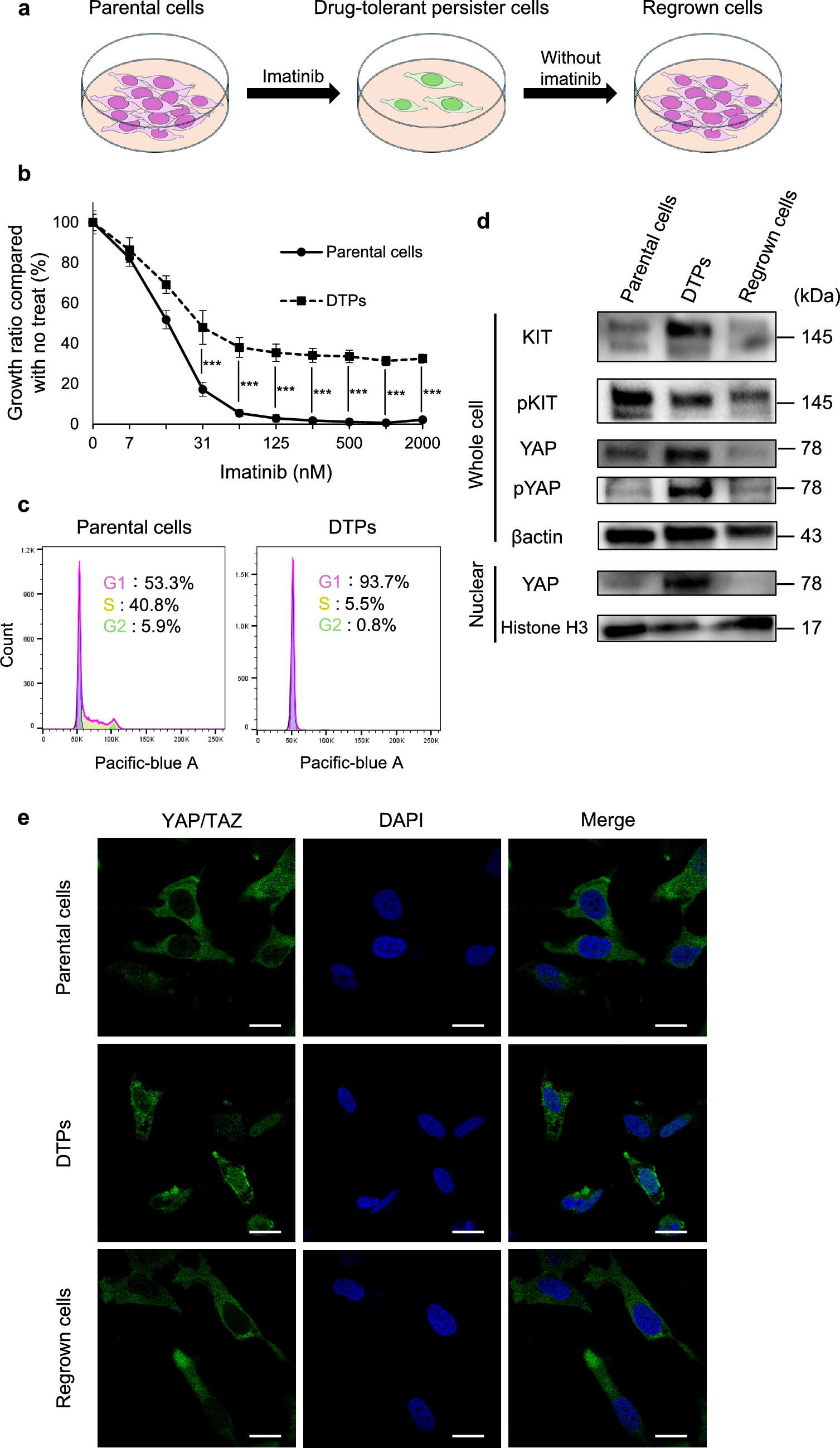


We performed immunohistochemical staining with an anti-CD8 antibody on samples from 157 cases of GC (Fig. 1A). The field of view was selected at the tumor margin to include the stroma in the tumor’s advanced region (Fig. 1B). Epstein–Barr virus (EBV)-positive cases were diagnosed using Epstein–Barr-encoded RNA in situ hybridization (Figure S1A–B). We created a histogram to illustrate the percentage of CD8 + TILs across 157 GC cases. Based on the distribution, the cases were divided into groups with 0%–10%, 11%–21%, and ≥ 22% CD8 + T-cell infiltration, which were classified as the LOW, MID, and HIGH groups, respectively (Fig. 1C). Of the 157 GC samples, 34 underwent scRNA-seq analysis and were categorized into 21 in the LOW group, 7 in the MID group, and 6 in the HIGH group (Fig. 1C). We investigated the correlation between the degree of CD8 + T-cell infiltration and clinicopathological features, revealing that the MID group was associated with an age < 67 years (p = 0.0235), a lower T stage (p = 0.0059), a lower N stage (p = 0.0072) and a lower Tumor, Node, Metastasis (TNM) stage (p = 0.0006) (Table S1). We examined the prognostic value of each group using Kaplan–Meier curves. The HIGH and LOW groups had significantly lower RFS (p = 0.0438) and OS (p = 0.0002) than the MID group, but no significant differences were observed between the HIGH and LOW groups (Fig. 1D–E). When EBV-positive cases were separated from other cases in the HIGH group, the RFS and OS of the EBV-positive HIGH group showed differences compared to the other HIGH groups, although these differences were not statistically significant. These findings are consistent with a trend toward a better prognosis in the EBV-positive group, as previously reported (Figures S1C–D) [11]. Additionally, we performed multivariate analyses based on clinicopathological characteristics (Table S2). Multivariate Cox regression analysis for sex, age, T stage, N stage, pathological stage, and the MID group versus the LOW and HIGH groups confirmed that pathological stage and the degree of CD8 + T-cell infiltration in the MID group were independent prognostic factors for OS (hazard ratio = 0.1306, 95% confidence interval: 0.04142–0.3912, p = 0.0003; and hazard ratio = 0.3581, 95% confidence interval: 0.1287–0.9182, p = 0.0377 respectively).
Fig. 1
Prognostic impact of the degree of CD8 + T-cell infiltration. A Representative immunohistochemistry (IHC) images of CD8 immunostaining in gastric cancer (GC) and adjacent tissues from CD8_IHC LOW, MID, and HIGH groups (from left to right). Scale bars, 100 μm. B Representative multiplex immunofluorescence images of CD8 + tumor-infiltrating lymphocytes (TILs) and epithelial cells using antibodies against CD8 (red) and AE1/AE3 (green), and 4',6-diamidino-2-phenylindole (DAPI) (light blue), in GC and adjacent tissues from the CD8_IHC LOW, MID, and HIGH groups (from left to right). Scale bars, 100 μm. C Histograms showing the percentage of CD8 + TILs across 157 GC cases (left) and 34 single-cell RNA sequencing (scRNA-seq) cases (right). The percentage of CD8 + TILs refers to the proportion of CD8 + TILs among total tumor cells within a field at 200 × magnification. The gray dotted line is the reference line separating the LOW, MID, and HIGH groups. D Recurrence-free survival analysis (performed using the Kaplan–Meier plotter) of patients with GC (n = 157; LOW group = 37, MID group = 81, and HIGH group = 39). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗∗ : p < 0.01, ∗ ∗ ∗: p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). E Overall survival analysis (performed using the Kaplan–Meier plotter) of patients (n = 157; LOW group = 37, MID group = 81, and HIGH group = 39). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001)
Variations in immune cell composition and IFNG expression within the GC TIMETo explore the differences in the TIME based on the degree of CD8 + T-cell infiltration, we performed scRNA-seq on 41 human samples, including 34 from GC tumor tissues and 7 from normal tissues (Fig. 2A). The clinical characteristics of the patients associated with these samples were recorded (Table S3). After quality control and normalization, our scRNA-seq dataset included 199,609 cells (Fig. S2A). We used data integration to reduce batch effects, performed principal coordinate analysis on the integrated data, and applied graph-based clustering to identify distinct cell populations (Fig. S2B–C). Based on the differentially expressed genes (DEGs) and canonical marker expression in each cluster, we identified 10 major cell types (Figs. S2D and 2B). The percentage of each immune cell was calculated among CD45-expressing immune cells, and higher numbers of T-cells and myeloid cells were observed in the tumors compared with the normal tissues (Fig. 2C).
Fig. 2
Variations in immune cell composition and IFNG expression within the GC tumor immune microenvironment. A Schematic illustration of the experimental workflow in our study. B Uniform manifold approximation and projection (UMAP) plot showing 10 color-coded major cell types, based on canonical marker genes. C Bar plots showing the proportions of seven immune cell clusters by the tissue source type. D Bar plots showing the proportions of seven immune cell clusters across four groups (HIGH, MID, LOW, and normal). E Scatter plot of CD8 + T-cell percentage and IFNG expression in GC cases. The regression equation and p value are displayed in the upper right corner. F Dot plot showing IFNG average expression in all cell clusters (left). The feature plot shows the IFNG average expression in UMAP (right). G Violin plots showing IFNG average expression in the CD8 + T-cells (top) and natural killer (NK) cells (bottom) across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). H Violin plots showing the IFNG gene set (IFNG.GS) score for all cell clusters
As shown in Fig. 1, we used CD8 staining of formalin-fixed paraffin-embedded specimens to classify the cases into HIGH, MID, and LOW groups based on CD8 + T-cell infiltration. The HIGH group showed a higher proportion of T-cells and myeloid cells compared with the MID group (Fig. 2D). Furthermore, after extracting all T-cells and NK cells and examining the proportions of CD4 + T-cells, CD8 + T-cells, and NK cells, we confirmed that the HIGH group had a higher proportion of CD8 + T-cells compared to the MID group, while the proportion of NK cells remained unchanged (Fig. S2E–F). We found that IFNG expression in the tumor environment correlated with the proportion of CD8 + TILs, even when excluding EBV-positive cases (Figs. 2E and S2G). IFNG was strongly expressed in T-cells and NK cells, mainly CD8 + TILs and NK cells (Figs. 2F and S2H), in which its expression increased stepwise across the groups (Fig. 2G). Interferon gamma (IFN-γ) is an inflammatory cytokine that activates T-cells and myeloid cells [12, 13]. Drawing from previous reports, we referenced a gene set influenced by the IFNG Gene Set (IFNG.GS) score to assess individual immune cell responsiveness to IFNG [13]. Among immune cells, the IFNG.GS score was high in T-cells, NK cells, myeloid cells, and B-cells (Fig. 2H). Overall, these results indicate that IFNG expression increases stepwise with the degree of CD8 + T-cell infiltration. Additionally, T-cells and myeloid cells, which comprise higher proportions of immune cells in the HIGH group, appear to be significantly associated with IFNG expression.
Heterogeneity in the abundance and functionality of CD8 + TILsTo elucidate the heterogeneity of CD8 + TILs and how their functions change in each environment, we extracted CD8 + T-cell clusters and analyzed them in detail. After re-clustering, we identified seven subclusters (Figs. S3A–C and 3A): progenitor-like T-cells (Prog_like), exhausted T-cells (Tex) expressing ITGAE (Tex_ITGAE), Tex expressing GZMK (Tex_GZMK), Tex expressing IGKC (Tex_IGKC), Tex expressing stress-related genes (TexSTR), Tex expressing interferon-stimulated genes (TexISG), and mucosal-associated invariant T-cell-like T-cells (MAIT_like), based on previously reported genetic markers [14, 15]. The proportions of the clusters did not vary significantly among the different groups (Fig. 3B).
Fig. 3
Heterogeneity in the abundance and functionality of CD8 + TILs. A UMAP plot showing seven color-coded subtype clusters of CD8 + TILs, based on representative genes. B Bar plots showing the proportions of seven subtype clusters of CD8 + TILs across four groups (HIGH, MID, LOW, and normal). C Violin plots showing IFNG.GS scores for the Tex_ITGAE subtype across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). D Violin plots showing the cytotoxic scores for the Tex_ITGAE subtype across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). E Violin plots showing the memory scores for the Tex_ITGAE subtype across four groups (HIGH, MID, LOW, and normal; left) and HIGH group (EBV-positive, EBV-negative, and normal; right). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). F Violin plots showing CD44 expression in the Tex_ITGAE subtype across four groups (HIGH, MID, LOW, and normal; left), and three HIGH groups (EBV-positive, EBV-negative, and normal; right). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). G Violin plots showing PDCD1 (left) and CTLA4 (right) expression in the Tex_ITGAE subtype across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). H Representative multiplex immunofluorescence images of PD1 + CD8 + TILs using antibodies against CD8 (red), PD1 (green), and DAPI (light blue), in GC and adjacent tissues from the MID group (right). Arrowheads indicate PD1 + CD8 + cells. Scale bars, 100 μm and 50 μm (low and high magnification, respectively). Quantification bar plot of PD1 + CD8 + cell counts across three groups (HIGH, MID, and LOW; left). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). I Violin plots showing CCL5 (left) and CXCL13 (right) expression in the Tex_ITGAE subtype across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). J Gene set enrichment analysis results comparing the HIGH and MID groups, showing enrichment of “GOBP_CELL_CYCLE”-associated gene sets in the Tex_ITGAE subtype for EBV-negative cases. K. Volcano plot comparing the HIGH and MID groups in the Tex_ITGAE subtype for EBV-negative cases. The red dots in the volcano plot represent genes with a p value < 0.05 and a fold change (FC) > 1.0
To investigate the functional state of the CD8 + TILs in antitumor immunity, we analyzed gene expression in the Tex_ITGAE subcluster, which has the highest cytotoxic score (Fig. S3D). The HIGH group showed higher IFNG expression than the MID group, but their CD8 + T-cells were less IFNG-responsive and had lower cytotoxicity and memory scores (Fig. 3C–D). However, memory scores were significantly higher in EBV-negative cases among the HIGH group (Fig. 3E). CD44, an activation marker, showed the lowest expression in the HIGH group (Fig. 3F) [16]. Similarly, PDCD1, an immune checkpoint gene, was downregulated in the HIGH group, whereas CTLA4 was upregulated (Fig. 3G). Within the HIGH group, the analysis was divided by EBV positivity, which did not affect the overall landscape of low PDCD1 and high CTLA4 expression (Fig. S3E). Additionally, analysis of TIGIT and LAG3 expression revealed that, compared to the MID group, the HIGH group showed higher expression of TIGIT and lower expression of LAG3 (Fig. S3F). Furthermore, immunofluorescence staining confirmed that the number of PD1 + CD8 + T-cells was lower in the HIGH group than the MID group (Fig. 3H).
We next used CellChat to determine which chemokines were the most potent CD8 + T-cell inducers in the TIME. We found that the CCL5 (RANTES) signal pathway was highly ranked, with the highest expression in the EBV-negative HIGH group (Fig. S3G–I and 3I). CCL5 is more effective at inducing T-cell chemotaxis than IFNG itself [17]. Furthermore, CXCL13 was most highly expressed in the EBV-negative HIGH group (Fig. 3I (Fig. S3I), whereas CXCR5 expression in B-cells was highest in the MID group (Fig. S3J). The CXCL13–CXCR5 axis is involved in ectopic or tertiary lymphoid structure (TLS) formation [18].
To understand the decreased CD8 + T-cell activity in the HIGH group, we conducted gene set enrichment analysis (GSEA). Upon excluding EBV-positive cases, the HIGH group had significantly lower expression of cell cycle-related genes than the MID group (normalized enrichment score = − 2.0084, p = 0.0003456, q = 0.1116) (Fig. 3J). Our volcano plot analysis revealed significant downregulation of Lck-interacting molecule 1 (LIME1), PTPRC-associated protein (PTPRCAP), and aldolase A (ALDOA) (Fig. 3K). Both LIME1 and PTPRCAP play important roles in TCR signaling and are required for T-cell activation [19, 20]. ALDOA is a glycolytic enzyme important in energy generation; it is involved in various cellular functions, such as cell shape maintenance and motility [21]. Thus, our data suggest that CD8 + TILs in the HIGH group recruit surrounding T-cells through chemokine signaling, thereby increasing cell count. However, these CD8 + T-cells show diminished cell division, downregulated TCR-related gene expression, and reduced glucose metabolism functionality.
TCR repertoire diversity in different groups of CD8 + TILsTo assess whether TCR signaling is dysfunctional in the HIGH group, we analyzed scTCR-seq data from seven samples coupled with our scRNA-seq data [22]: two each from the HIGH, MID, and LOW groups, and one control sample (Table S3). None of the cases were EBV-positive. First, we examined the total number and percentage of unique clones to characterize the TCR sequences into clusters of CD8 + T-cells (Fig. 4A–B). The total number of unique clones was high in the Prog_like, Tex_ITGAE, and Tex_GZMK groups, with the highest number of unique clones in Tex_GZMK. The percentage of unique clones was low in the Tex_ITGAE and Tex_GZMK groups, which suggests that clonality is high. Therefore, we visualized the number of clones within each cluster across four groups (HIGH, MID, LOW, and normal) to identify the groups in which the clones were expanding (Fig. 4C). The MID group had the most clones, which were widely distributed in the Prog_like, Tex_ITGAE, and Tex_GZMK clusters. Additionally, by employing the clonalOverlay function to visualize the clonality transition within each cluster across different groups [23], we discovered that the Tex_ITGAE cluster exhibited lower clonal expansion in the HIGH group than in the MID group (Fig. 4D). The occupied CD8 + T-cell repertoire spaces showed greater clonality in the MID group than in the HIGH and LOW groups (Fig. 4E). This was also observed for the Tex_ITGAE and Tex_GZMK clusters (Fig. 4F). Finally, we examined the relative number of clones shared in each cluster and found that shared clones in Prog_like, Tex_ITGAE, and Tex_GZMK accounted for the majority (Fig. 4G). Briefly, TCR repertoire analysis showed decreased clonality in the HIGH group compared with the MID group.
Fig. 4
TCR repertoire diversity of different groups of CD8 + TILs. A Bar plots showing the unique clone counts of seven subtype clusters of CD8 + TILs across four groups (HIGH, MID, LOW, and normal). B Bar plots showing the percentage of unique clones within seven subtype clusters of CD8 + TILs across four groups (HIGH, MID, LOW, and normal). C UMAP plot showing the clone size of the CD8 + TILs across four groups (HIGH, MID, LOW, and normal). The UMAP plot shows the number of cells used for single-cell T-cell receptor sequencing (scTCR-seq): 471 cells in the HIGH group, 2063 cells in the MID group, 623 cells in the LOW group, and 203 cells in the normal group. D UMAP contour plot showing the clone frequency distribution of the CD8 + TILs across four groups (HIGH, MID, LOW, and normal). The background UMAP plot shows the number of cells used for scRNA-seq: 6477 cells in the HIGH group, 5359 cells in the MID group, 13,785 cells in the LOW group, and 4746 cells in the normal group. E Bar plots showing the proportions of occupied repertoire spaces of CD8 + TILs across four groups (HIGH, MID, LOW, and normal). F Bar plots showing the proportions of occupied repertoire spaces for the Tex_ITGAE (top) and Tex_GZMK (bottom) subtypes across four groups (HIGH, MID, LOW, and normal). G Chord diagram showing the clonal relationships between seven subtype clusters of CD8 + TILs
Characterization of myeloid cell dynamics and pathway interactionsWe next analyzed myeloid cell clusters in detail. As a result of re-clustering, 10 subclusters were identified (Figs. S4A–B and 5A): dendritic cells (DCs) expressing CCR7 (DC_CCR7), DCs expressing CD1C (DC_CD1C), DCs expressing CLEC9A (DC_CLEC9A), macrophages expressing APOE (M_APOE), macrophages expressing FCN1 (M_FCN1), myeloid-derived suppressor cells (MDSCs) expressing OLR1 (MDSC_OLR1), neutrophils expressing CXCL8 (N_CXCL8), neutrophils expressing IGKC (N_IGKC), neutrophils expressing S100A8 (N_S100A8), and plasmacytoid DCs (pDC), as described in previous reports [24,25,26]. We observed increased percentages of neutrophils and MDSCs in the tumor and in the HIGH group compared with the normal tissue and MID group, respectively, whereas the DC_CLEC9A percentages remained unchanged (Fig. 5B). Based on previous reports, we identified DC_CLEC9A as conventional type 1 DCs (cDC1s) that present antigens to CD8 + T-cells [24]. HIGH group cDC1s showed reduced IFNG-responsiveness, activity, and cross-presentation scores compared with MID group cDC1s, with a significant decrease in EBV-negative cases (Figs. S4C–D and 5C–D). Immunofluorescence staining revealed the highest number of cDC1s in the tumor margins of the MID group, with close proximity to CD8 + cells (Fig. S4E and 5E–F). Analysis of immunosuppressive cells showed that M2-like macrophages (M_APOE) had a higher immunosuppression score in the LOW group than in the MID and HIGH groups (Fig. 5G). Conversely, MDSC_OLR1 had the highest immunosuppression score in the HIGH group, regardless of EBV positivity (Fig. 5H).
Fig. 5
Characterization of the dynamics and pathway interactions of myeloid cells. A UMAP plot showing 10 color-coded subtype clusters of myeloid cells based on representative genes. B Bar plots showing the proportions of 10 subtype clusters of myeloid cells by the tissue source type (left) and in four groups (HIGH, MID, LOW, and normal; right). C Violin plots showing the activation scores (left) and cross-presentation scores (right) for the DC_CLEC9A subtype across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). D Violin plots showing the cross presentation scores of the DC_CLEC9A subtype across three HIGH groups (EBV-positive, EBV-negative, and normal; right). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). E Representative multiplex immunofluorescence images of CD8 + and CLEC9A + cells using antibodies against CD8 (red) and CLEC9A (green), and DAPI (light blue), in GC and adjacent tissues from the MID group. Arrowheads indicate CLEC9A + cells. Scale bars, 100 μm and 50 μm (low and high magnification, respectively).Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). F Quantification bar plot of the CLEC9A + cell counts across three groups (HIGH, MID, and LOW). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). G Violin plots showing the suppression scores of the M_APOE subtype across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). H Violin plots showing the immunosuppressive scores of the MDSC_OLR1 subtype across four groups (HIGH, MID, LOW, and normal; left) and three HIGH groups (EBV-positive, EBV-negative, and normal; right). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). I Chord diagram showing the secreted signaling pathways of GALECTIN (left) and RESISTIN (right) among eight subtype clusters: M_APOE, MDSC_OLR1, DC_CLEC9A, DC_CCR7, eTreg, Tex_ITGAE, Tex_GZMK, and epithelial cells, using CellChat. J Violin plots showing RETN expression in the MDSC_OLR1 subtype across four groups (HIGH, MID, LOW, and normal; left) and three HIGH groups (EBV-positive, EBV-negative, and normal; right). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). K Dot plots showing the secreted signaling pathways (MIF and RESISTIN) in MDSC_OLR1 and epithelial cells across three groups (HIGH, MID, and LOW) using CellChat. L Violin plots showing LGALS9 expression in the DC_CCR7 (left) and epithelial cells (right) across four groups (HIGH, MID, LOW, and normal). Statistical significance was set at p < 0.05 (ns: p ≥ 0.05, ∗ : p < 0.05, ∗ ∗ : p < 0.01, ∗ ∗ ∗ : p < 0.001, and ∗ ∗ ∗ ∗ : p < 0.0001). M. Dot plots showing the secreted signaling pathways (GALECTIN) from DC_CCR7, MDSC_OLR1, and epithelial cells across three groups (HIGH, MID, and LOW) using CellChat
Analysis of the secreted signaling pathways between cell types in the HIGH group using CellChat revealed that the GALECTIN, RESISTIN, and MIF pathways were ranked the highest (Fig. S3G). The GALECTIN pathway was primarily sourced from myeloid cells and epithelial cells, whereas the RESISTIN pathway was sourced only from MDSCs (Fig. 5I). The RESISTIN pathway, involved in chronic inflammation by binding to CAP1 in monocytes and upregulating inflammatory cytokines, is primarily expressed in MDSCs (Fig. S4F) [27]. It is most highly expressed in the EBV-negative HIGH group, with strong interactions with DC_CCR7, M_APOE, and CD8 + TILs (Fig. 5J–K). Additionally, among the three groups, the HIGH group exhibited enhanced MIF–CD74 pathway signaling, which upregulates CXCL8 in T-cells and induces neutrophils (Fig. 5K) [28]. Galectin, a β-galactoside-binding lectin, binds to glycans on cell membranes and inhibits TCR and BCR signaling via CD45 [29, 30]. Galectin-9 binds to CD44, activates Smad3 through TGF-β receptor complexes, and enhances Treg cell stability and function [31]. LGALS9 is highly expressed in DC_CCR7 and epithelial cells in the HIGH group, suggesting a role in T-cell immunosuppression (Fig. 5L). Cells in the DC_CCR7 subcluster, which highly express CCR7 and LAMP3, are recognized as mature dendritic cells enriched in immunoregulatory molecules (mregDCs) [24]. Our analysis showed enhanced LGALS9–CD45 signaling (from mregDCs and epithelial cells to CD8 + T-cells) and LGALS9-CD44 signaling (to effector Tregs [eTregs]) in the HIGH group (Fig. 5M).
Thus, the HIGH group had lower expression of markers associated with cDC1 antigen-presenting capacity and higher expression of those associated with the suppressive function of MDSCs than the LOW and MID groups. The GALECTIN pathway was identified as a candidate factor for the suppression of TCR signaling in CD8 + TILs.
Analysis of eTregs and CTLA4 expression across different CD8 + TIL groupsAfter re-clustering of the Treg clusters, four subclusters were identified (Figs. S5A–C and 6A): progenitor-like Tregs (Prog_like), activated Tregs (activatedTreg), eTregs, and Tregs expressing interferon-stimulated genes (TregISG), as in previous reports [32]. The proportion of each cluster did not change significantly in the HIGH group compared with the MID group (Fig. 6B). As with CD8 + TILs, IFNG responsiveness was reduced in the HIGH group compared with the MID group, and the HIGH group had a lower Treg suppression score, but a high memory score (Fig. 6C–D). As in previous reports [32], CTLA4 expression was highest in eTregs (Fig. S5D). CD44 expression was low in the HIGH group, but CTLA4 showed high expression, similar to the MID group (Fig. 6E–F). Furthermore, CTLA4 expression tended to be higher in the EBV-negative HIGH group (Fig. 6F). The IL2 receptor is essential for regulating Treg homeostasis and suppressive activity [33]. IL2RA expression in eTregs did not differ significantly among the three groups (Fig. 6G). The chemokine receptor CXCR3, induced by IFN, promotes the migration of T-cells into inflammatory sites and the tumor microenvironment. CXCR3-expressing Tregs are abundant in tumors and inhibit CD8 + TIL activity by preferentially interacting with cDC1 [34]. CXCR3 expression was highest in eTregs and maintained in the HIGH group, although at a lower level than in the MID group (Fig. S5E and 5G). Immunofluorescence staining showed a higher percentage of CTLA4 + Tregs in the HIGH group than the MID group, although the number of Tregs themselves was unchanged (Fig. 6H–I). Thus, although Treg activity was slightly lower in the HIGH group than in the MID group, the expression of the inhibitory receptor CTLA4 was higher, suggesting that it acts to suppress antitumor immunity.
Fig. 6
Analysis of Treg subclusters and CTLA4 expression across different CD8 + TIL groups. A UMAP plot sho
Comments (0)