
Remember me

Between April 2018 and July 2020, 91 individuals with hematologic malignancies were screened for eligibility in the SMARTrial (Fig. 1). Eighty individuals (88%) fulfilled the eligibility criteria and were enrolled (Fig. 2a). The primary endpoint, ‘successful completion of the ex vivo drug response assay within 7 d’, could be assessed in all 80 individuals. For the secondary endpoint analysis, to systematically correlate ex vivo and in vivo drug responses, we assigned participants to two major subcohorts. Cohort 1 included individuals treated with chemotherapy (n = 46), and cohort 2 included individuals treated with venetoclax or ibrutinib (n = 18). Sixteen individuals were excluded from the secondary endpoint analysis because in vivo response to the prescribed treatment could not be assessed according to the study protocol (n = 9), ex vivo response profiles did not pass quality control (n = 2), or participants could not be assigned to either of the two subcohorts due to their prescribed therapies (n = 5).
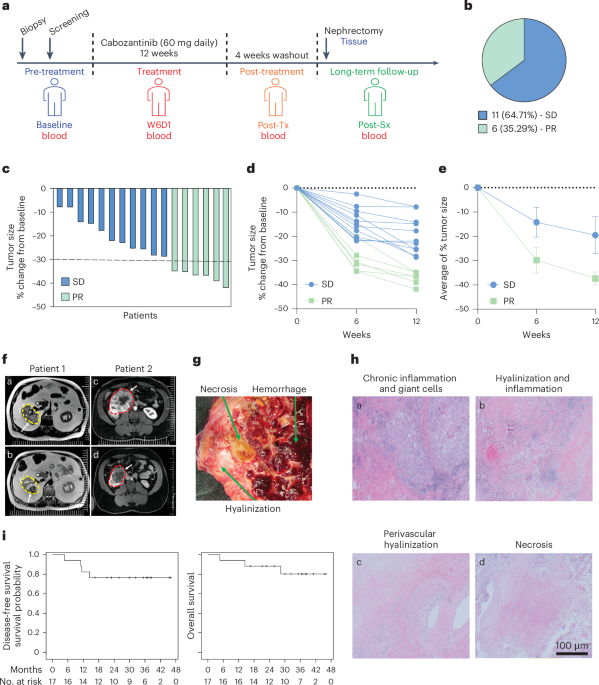
Fig. 1: Overview of study design and drug selection.
a, Outline of the prospective observational study. b, Overview of the drug classes in the compound library based on their targets/mode of action. FDA, Food and Drug Administration; TNF, tumor necrosis factor; IDH, isocitrate dehydrogenase; PLK, polo-like kinase 1; TLR, Toll-like receptor; HGF, hepatocyte growth factor; PKC, protein kinase C.
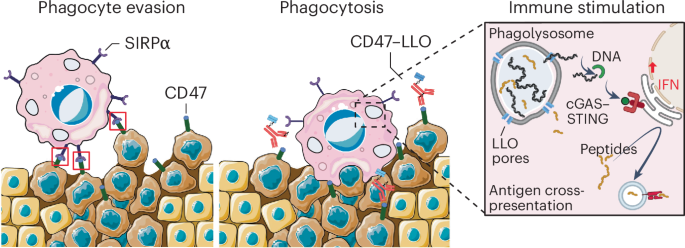
Fig. 2: Flow diagram and primary endpoint analysis.
a, Flow diagram of study participants and downstream analyses. The asterisk indicates that in nine individuals, the in vivo response was not evaluable due to the following reasons: scheduled treatment could not be initiated due to infection leading to death (n = 1), fulminant PD leading to early death (n = 1), scheduled treatment was denied by the participant before the start of treatment (n = 1) or within 14 d after treatment start (n = 1), scheduled treatment was prematurely discontinued due to side effects without prior response assessment (n = 2), scheduled treatment was discontinued several times due to side effects (n = 1) or response assessment was not available (n = 2). The section sign (§) indicates that five participants could not be assigned to the two subgroups (chemotherapy cohort and CLL cohort treated with venetoclax or ibrutinib) used for the secondary endpoint analysis. These participants were treated with palbociclib (n = 1), alemtuzumab (n = 1) and idelalisib ± rituximab (n = 2). One participant was treated with ibrutinib and rituximab but had a diffuse large B cell lymphoma. b, Enrolled participant cohort by diagnosis. c, Rate of successfully completed drug response assessments within 7 d (primary endpoint). d, Subcohorts for secondary endpoint analysis. MCL, mantle cell lymphoma; DLBCL, diffuse large B cell lymphoma; B-PLL, B cell prolymphocytic leukemia; T-NHL, T cell non-Hodgkin lymphoma.
Demographics and disease characteristics for all 80 eligible participants are shown in Table 1 and Supplementary Table 1. The median age was 68.5 years (range of 20–91 years). Participants with the following hematologic malignancies were included: AML (34/80, 42%), ALL (2/80, 2%), CLL (25/80, 31%), aggressive T cell leukemia (T cell prolymphocytic leukemia (T-PLL) 4/80, 5%), aggressive B cell lymphoma (5/80, 6%), aggressive T cell lymphoma (1/80, 1%) and indolent B cell lymphoma (9/80, 11%; Fig. 2b). The majority of participants were treatment naive (54/80, 68%), but individual participants were heavily pretreated with three or more prior lines of therapy before study entry (6/80, 8%). The median follow-up time of the study cohort was 2.1 years. Median time from diagnosis or determination of treatment indication to treatment initiation ranged from 2 d in T-PLL to 32 d in follicular lymphoma (FL; Supplementary Table 2).
Table 1 SMARTrial participant characteristicsHuman-derived primary tumor cells were obtained from peripheral blood (59/80, 74%), bone marrow aspirates (12/80, 15%) and lymph node biopsies (9/80, 11%). The median tumor purity of all 80 samples was 84.5% (range, 52–99%) and was assessed by immunophenotyping, peripheral blood smears or bone marrow cytology.
Components of almost all in vivo therapies (95%) recommended by the treating physician were included in our diagnostic ex vivo drug test. Sixty-four percent of participants (51/80) were scheduled for chemotherapy at study entry, either alone or in combination with immunotherapies and small-molecule inhibitors (Supplementary Table 3). Chemotherapy-free treatments, such as ibrutinib or venetoclax, were intended in 36% of participants (29/80). In total, three participants did not start the scheduled treatment due to fulminant progressive disease (PD), infection leading to death and refusal of the therapy by the participant after study inclusion.
Feasibility of ex vivo DRP for clinical decision-makingThe primary objective of the non-interventional SMARTrial was to evaluate the feasibility of a short-term ex vivo DRP assay for primary human-derived cancer cells in the clinical routine. Therefore, we evaluated the rate of successfully completed drug response assays within 7 d as the primary endpoint. The primary endpoint was met in 91.3% (95% confidence interval (95% CI) of 82.8–96.4%) of all eligible participants (Fig. 2c). The median time until the release of the final drug response report was 3 d (interquartile range (IQR) of 2–6 d, range of 2–17 d). The DRP was reported in the ‘SMARTrial explorer’, an interactive web application (http://mozi.embl.de/public/SMARTrial/), which could be accessed by the treating physicians. Reasons for delayed reporting in individual participants included suboptimal sample quality or study enrollment of participants before public holidays.
Quality assessmentWe performed several steps of data quality assessment before the ex vivo DRP data were used for further exploratory analyses. First, we estimated the technical variability by calculating the standard deviation (s.d.) of 16 evenly distributed dimethyl sulfoxide (DMSO) controls for each drug plate per participant (Extended Data Fig. 1a). In general, the median s.d. was low (median s.d.: 0.08; range: 0.03–0.6), and we found no significant difference between the samples derived from different tumor sample origins (peripheral blood, bone marrow and lymph node; P = 0.86, one-way analysis of variance). Four myeloid and three lymphoma plates belonging to a total of four participants (S047, S050, S056 and S062) showed relatively high technical noise (s.d. of negative controls > 0.3). For two participants (S047 and S062), no additional tumor material was available for retesting, and these samples were therefore excluded.
In addition, we assessed the validity of our ex vivo DRP pipeline by analyzing if expected drug–drug correlations and known gene–drug associations are recapitulated. Drugs with similar modes of action (for example, BTK inhibitors, BCL-2 inhibitors and vinca alkaloids) strongly correlated with each other (Extended Data Figs. 1b and 2), and participant samples clustered by diagnosis (Extended Data Figs. 3 and 4). Clinically well-established therapeutics recapitulated known vulnerabilities conferred by mutations, such as in FLT3 and IDH1, in AML cells. For example, tumor cells with mutations in the FLT3 tyrosine kinase domain (FLT3-TKD) were sensitive to the type I FLT3 inhibitors crenolanib, gilteritinib and midostaurin, which bind the FLT3 receptor in the active conformation and are known to be active in AML cells with mutations in the FLT3 internal tandem duplication (FLT3-ITD) and FLT3-TKD. By contrast, these tumor cells were insensitive to the type II FLT3 inhibitors quizartinib and sorafenib, which are known to be inactive in FLT3-TKD-mutated AML cells13 (Extended Data Fig. 5a,b). IDH1-mutated AML cells were specifically sensitive to venetoclax, which confirmed the known dependency of IDH-mutated AML on BCL-2 (ref. 14; Extended Data Fig. 5c). TP53-mutated tumor cells showed a decreased sensitivity to Nutlin-3a compared to wild-type tumor cells10,15,16 (Extended Data Fig. 5d). Drug responses can be explored in the interactive web application (http://mozi.embl.de/public/SMARTrial/).
Association between ex vivo and in vivo responsesAn important question of our study was to understand if ex vivo drug responses and in vivo responses correlate with each other. Considering the ongoing relevance of chemotherapy for hematologic tumors but also the increasing importance of chemotherapy-free targeted treatments in blood cancer, we focused on two subgroups: participants treated with chemotherapy (n = 46) and participants with CLL treated with venetoclax or ibrutinib (n = 18).
Among the 46 participants in the chemotherapy cohort, 29 were diagnosed with AML, 2 were diagnosed with ALL, and the remaining 15 were diagnosed with B or T cell lymphoma. All participants were treated with standard cytotoxic chemotherapy regimens combined with a targeted therapy (monoclonal antibodies or small-molecule inhibitors) in 46% of treatments.
Our ex vivo drug screen covered a broad library of compounds with different modes of action. To determine which ex vivo drug response profiles were most suitable for predicting chemosensitivity in vivo, we chose an unbiased approach and associated all ex vivo drug response profiles with in vivo response categories (PD, stable disease (SD) and response (R)). Because direct comparisons between drugs were not feasible due to small numbers of uniformly treated participants and combination therapies in vivo, we grouped drugs according to their mode of action and associated the averaged ex vivo responses across all drugs within these classes with the in vivo response groups (R versus PD; Extended Data Fig. 5e). Ex vivo responses between chemosensitive and chemorefractory individuals differed most significantly for the following five drug classes: heat shock protein inhibitors (stress response), cyclin-dependent kinase inhibitors, proteasome inhibitors, chemotherapeutics and inhibitors involved in the DNA damage response signaling pathway. In a second step, we associated ex vivo drug response profiles of each individual drug with in vivo response (R versus PD; Extended Data Fig. 5f). Multiple drugs of the above-mentioned drug classes showed similar activity, suggesting that our data represent on-target effects as the primary mode of action of these drugs. Additional drugs with significantly different ex vivo drug sensitivities between responders and participants with PD were found among the groups of mTOR inhibitors, proteasome inhibitors and compounds that are involved in histone modifications. Representative examples of dose–response curves and averaged drug viabilities between individuals who were chemosensitive and chemorefractory are shown in Fig. 3a.
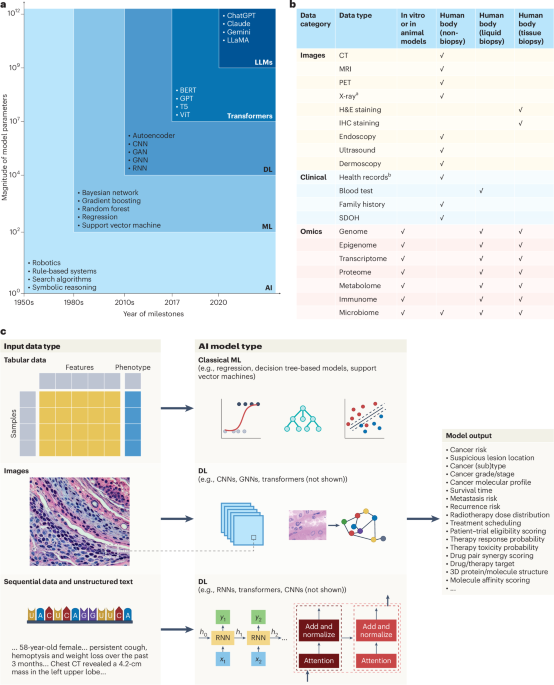
Fig. 3: Association between ex vivo drug response and in vivo response or clinical outcome.
a, Ex vivo sensitivity by clinical response group. Dose–response curves built by fitting a five-parameter logistic model using ex vivo viability measurements. Individual participant observations are displayed by circles in both plots. Blue and red represent groups of participants with clinical response (R) versus participants with PD, respectively (R: n = 33; PD: n = 5). Error bars represent mean and 95% CI. Centers, hinges and whiskers of the box plots signify medians, quartiles and 1.5× IQR, respectively. b, Elastic net logistic regression model of ex vivo drug viability (AUC) to chemotherapeutic agents with binary endpoint R versus PD (R: n = 33; PD: n = 5). The median odds ratio (OR) presented here relates to a change in ex vivo drug viability of 10%. Covariates are shown ordered by selection proportion (>0.5 shown here). The results of all covariates included in the model are shown in Supplementary Table 2. c, Association of ex vivo drug responses and EFS assessed by univariate Cox regressions (R: n = 33; SD: n = 5; PD: n = 5). Estimated hazard ratios with corresponding 95% CIs are shown. Ex vivo drug viability (AUC) was calculated per drug and scaled such that a unit change of the regressor corresponds to a 10% change in cell viability. P values are from two-sided Wald tests on Cox regression models. d, Kaplan–Meier plots for EFS stratified by ex vivo drug response to vincristine and vindesine (R: n = 33; SD: n = 5; PD: n = 5). Participant groups of ex vivo responders and weak responders were defined by ex vivo drug responses dichotomized using maximally selected log-rank statistics to visualize effects. Fourteen of 43 participants were classified as vincristine weak responders, and 15 of 43 participants were classified as vindesine weak responders.
Individual ex vivo drug responses might not predict overall chemosensitivity. Therefore, we investigated how the combination of multiple ex vivo drug responses could be used to predict chemosensitivity. Because the majority of relevant drugs with significant differences were found among the chemotherapeutics (the treatment most individuals received in vivo), we focused on the group of chemotherapeutics to avoid overfitting. For variable selection, we built an elastic net logistic regression model and regressed ex vivo drug response profiles of individual chemotherapeutic agents to in vivo response (R versus PD; Fig. 3b and Supplementary Table 4). In total, we fitted 1,000 models based on different randomly selected folds. In more than 80% of all models, both vinca alkaloids, vincristine and vindesine, the two anthracyclines idarubicin and mitoxantrone and the purine analog cladribine were selected as prognostic features. Our models including the five above-mentioned chemotherapeutics reached a median cross-validation area under the receiver operating characteristic curve (AUROC) of 0.84 to 0.85, highlighting the discriminative ability of these models.
To elucidate the predictive power of features selected by the elastic net regression model on the durability of achieved clinical responses, we regressed event-free survival (EFS) on these drug response profiles. An event was defined as PD, change of treatment or death. We found that stronger ex vivo responses to both vinca alkaloids were associated with extended EFS in the chemotherapy cohort (Fig. 3c,d). Together, these data suggest that ex vivo drug response phenotyping is useful in predicting important clinical endpoints in individuals treated with chemotherapy across hematologic malignancies.
We further investigated if the tumor cell infiltration across the chemotherapy cohort, which ranged from 54 to 97%, had an impact on the observed ex vivo response to chemotherapeutic agents and may have confounded the ex vivo–in vivo drug response association. We observed a weak correlation between tumor cell infiltration and ex vivo response to chemotherapeutic agents (r = −0.33, P = 0.02; Extended Data Fig. 6a) but no correlation between tumor cell infiltration and in vivo response (Extended Data Fig. 6b). Furthermore, we used tumor infiltration as a blocking factor that did not affect the significance of most ex vivo–in vivo drug response associations (Extended Data Fig. 6c). We conclude that ex vivo–in vivo drug response associations for chemotherapeutic agents are not confounded by tumor infiltration rate in our study.
Although this study was a non-interventional study and individuals were treated according to the treatment that was scheduled by the treating physician before study entry, one participant (S005) received ex vivo drug response-informed treatment after failure of all standard chemotherapies. This participant suffered from refractory Burkitt lymphoma (BL). Lymphoma cells were insensitive to almost all drugs in the drug screen (Extended Data Fig. 7a). However, in the drug ranking, we identified a strong ex vivo sensitivity to pralatrexate. This suggested that the participant might benefit from treatment with a folate antagonist. The participant agreed to this individual ex vivo-guided treatment approach, and after three cycles of high-dose methotrexate, the participant achieved a partial response. Subsequently, the participant underwent a consolidating allogeneic stem cell transplantation, which resulted in complete remission (Extended Data Fig. 7b). This example illustrates how ex vivo DRP can reveal unexpected effective anticancer drugs and support treatment decisions, especially in individuals for whom standard treatments are no longer available.
We further investigated the association between ex vivo and in vivo responses of targeted therapies using the example of individuals with CLL. Our study cohort included eight individuals who were treated with venetoclax and ten individuals who were treated with ibrutinib. Venetoclax was combined with an anti-CD20 treatment in the majority of participants (seven of eight). Concordant with the in vivo response, the primary tumor cells of all participants who were treated with venetoclax showed ex vivo sensitivity to venetoclax (Extended Data Fig. 7c). Ibrutinib exhibited smaller effect sizes than venetoclax in our short-term ex vivo assay, which is in line with clinical response dynamics and previous studies10. Cells from the only participant who showed insufficient in vivo efficacy of ibrutinib exhibited the weakest ex vivo response to ibrutinib (S069; Extended Data Fig. 7d). This participant had received ibrutinib before, which was discontinued due to atrial fibrillation. A genetic profiling of this participant’s tumor cells was performed, but no mutation known to confer ibrutinib resistance was found, demonstrating the potential to improve response prediction beyond known genetic risk markers.
Validation of ex vivo and in vivo response associationWe further aimed to investigate if ex vivo DRP may improve clinical standard genetic risk profiling. Therefore, we focused on first-line treatment of AML, where genetic risk profiling is considered clinical standard. We assembled a validation cohort of 95 clinically well-annotated AML biosamples from the AML biobank of the German Study Alliance for Acute Myeloid Leukemia (SAL). All biosamples were obtained from treatment-naive participants with AML who were scheduled to receive induction therapy with daunorubicin and cytarabine. The cohort was compiled to contain 47 responders and 48 non-responders to induction therapy from all ELN-22 risk groups (Fig. 4a, Table 2 and Supplementary Table 5).
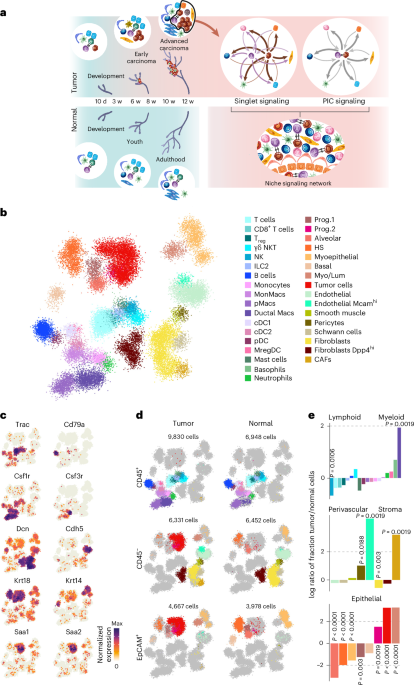
Fig. 4: Validation of the association between ex vivo and in vivo drug responses in a cohort of individuals with AML.
a, Overview of the validation cohort. The inner circle represents ELN-22 risk groups in total numbers, and the outer circle represents the distribution of in vivo responders and non-responders in ELN-22 risk groups. b, Ex vivo treatments with significantly different responses in in vivo responders (n = 47) and non-responders (n = 48). Negative log10 (P value) of Student’s t-tests is shown on the y axis, and mean difference between responders and non-responders is on the x axis. The dashed line represents the 10% false discovery rate cutoff (Benjamini–Hochberg procedure); NS, not significant. c, Viability (AUC) after ex vivo treatment with vincristine (top) and viability (volume under the curve (VUC)) after treatment with daunorubicin and cytarabine (bottom) separated by ELN-22 risk groups (ELN-22 adverse risk: non-responder: n = 28, responder: n = 8; ELN-22 intermediate risk: non-responder: n = 14, responder: n = 29; ELN-22 favorable risk: non-responder: n = 6, responder: n = 10). P values are derived from two-sided Student’s t-tests. Centers, hinges and whiskers of the box plots signify medians, quartiles and 1.5× IQR, respectively. d, Kaplan–Meier plots for EFS stratified by ex vivo drug response to vincristine and daunorubicin + cytarabine. For visualization purposes, participant groups of ex vivo responders and weak responders were defined by ex vivo drug responses dichotomized using maximally selected log-rank statistics to visualize effects. Sixty-four of 95 participants were classified as vincristine weak responders. Sixty of 95 participants were classified as daunorubicin + cytarabine weak responders. P values are from two-sided Wald tests on Cox regression models using drug responses as continuous variables. e, Forest plot of hazard ratios in multivariate Cox proportional hazards models for EFS including ELN-22 risk groups and viability after ex vivo treatment with vincristine (top) and daunorubicin + cytarabine (bottom). The ex vivo responses (AUC values) were centered by mean and scaled by 2 s.d. to bring them to a similar scale as the categorical ELN-22 risk group variables.
Table 2 Validation cohort participant characteristicsAs a first step, we compared ex vivo drug response in the validation cohort between clinical responders and non-responders independently of genetic risk profiles. Ex vivo drug response profiles differed significantly between in vivo responders and non-responders (Benjamini–Hochberg-adjusted17P value of <0.1). Interestingly, clinical in vivo response to induction therapy with daunorubicin and cytarabine was significantly associated with ex vivo response to the combination treatment with daunorubicin and cytarabine but not with ex vivo response to either drug alone. Vincristine, vindesine and cladribine, which were among the strongest predictors of in vivo response in the elastic net logistic regression model in the SMARTrial cohort, again showed the strongest association between in vivo and ex vivo response in the validation cohort (Fig. 4b and Extended Data Fig. 8).
As a next step, we investigated whether ex vivo DRP could further improve clinical in vivo response prediction and compared drug response profiles within each ELN-22 risk group, a well-established and very recently updated AML risk stratification tool4. Ex vivo response profiles significantly distinguished in vivo responders to daunorubicin and cytarabine from non-responders in the genetic adverse risk group defined as per ELN-22 recommendations4 (Fig. 4c and Extended Data Fig. 9).
In addition, we regressed ex vivo drug response profiles on EFS to assess the ability of ex vivo drug response to predict durability of achieved responses. Indeed, poor ex vivo drug response to vincristine as well as daunorubicin and cytarabine was associated with adverse EFS in participants with AML after induction therapy with daunorubicin and cytarabine (Fig. 4d). Ex vivo DRP further improved outcome prediction especially in participants with adverse risk as determined by ELN-22 (Fig. 4e and Extended Data Fig. 10). These results suggest that ex vivo DRP may improve in vivo response prediction beyond established genetic risk stratification tools in AML.
Comments (0)